生物信息bowtie比对软件的结果格式说明
生物信息分析中会用到很多的比对软件,比较常用的有bowtie、bowtie2、bwa等,比对文件的标准格式是sam格式,但是bowtie比对默认输出的格式却不是sam格式,由于bowtie适用于短序列比对,并且看突变碱基比较方便,因此它的默认输出格式还是有一定优势的,下面就来说明一下它的默认输出格式。
example:
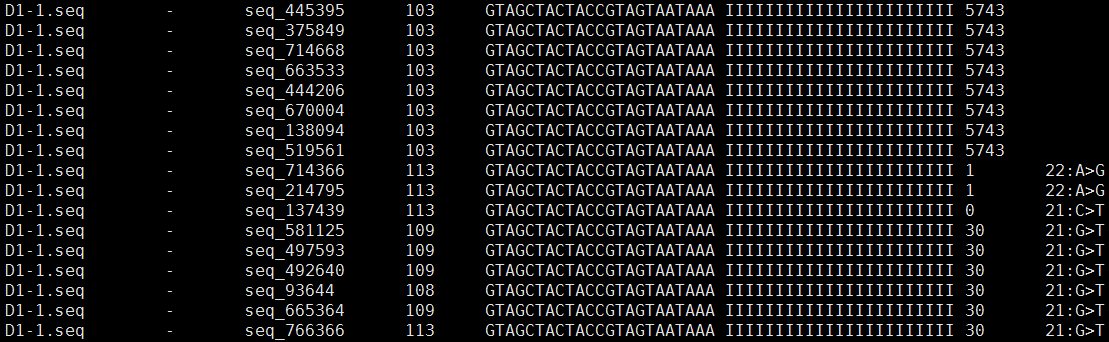
说明:
1. query id,也就是你自己用于比对的序列的ID。
2. + 表示正向比对上这条reads;- 表示对反向比对上这条reads。
3. 对应比对上的参考序列的ID,ref。
4. 自己用于比对的reads比对到参考序列的起始位置,注意下标是从0开始计数,例如10代表从第11个碱基开始比对上。第2列为"+"时,表示query第一个碱基map到reference上的位置;第2列为"-"时,表示query的反向互补序列第一个碱基map到reference上的位置。
5. 如果第2列为"+",则和query序列一致;否则,和query序列反向互补。
6. 质量文件,如果query文件为fasta格式,则无法获取质量文件,用I代替,I的数量与query序列长度一致。
7. 当前query能map到GENOME的4个不同位置
8. 如果存在第8列,表示有错配,bowtie的mismatch最大可设置为3。第8列可以分为三个部分,最左端的数字,中间的碱基为reference碱基,最右端的碱基为query碱基,下面分情况讨论:
第2列为"+"时:最左端的数字9表示query从5'端数起,第10个碱基为"T",而对应的reference为"G";
第2列为"-"时:最左端的数字9表示query先作反向互补,然后从3'端数起,第10个碱基为"T",而对应的reference为"G";

 浙公网安备 33010602011771号
浙公网安备 33010602011771号