
EDTA安装及使用
EDTA,该软件包用于全基因组TE的从头注释。EDTA设计目的是为全基因组生成一个高质量、无冗余的TE库,并过滤掉原始候选TE中的错误发现。EDTA官网
EDTA整合了多种TE注释软件,具体框架结构以及包含的软件如下:
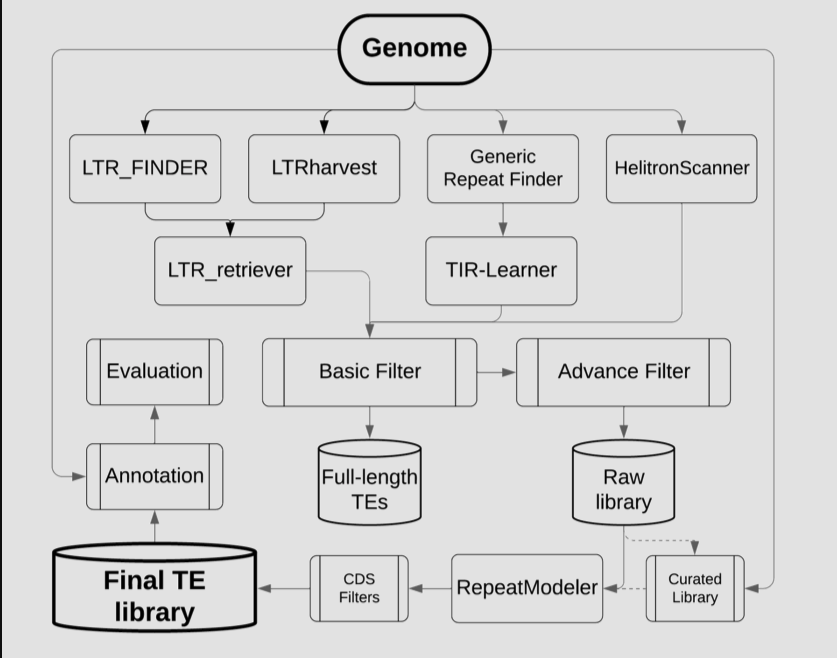
一、安装
#途径一(mamba很好用,但是我这里安装总出错)
conda create -n EDTA
conda activate EDTA
conda install -c conda-forge -c bioconda mamba
mamba install -c conda-forge -c bioconda edta python=3.6 tensorflow=1.14 'h5py<3'#途径二(亲测好用)
git clone https://github.com/oushujun/EDTA.git
cd EDTA
conda env create -f EDTA.yml二、使用
#每次使用别忘了激活EDTA环境,其中有EDTA运行必备的依赖包;同时,记得切换至EDTA目录下或者使用EDTA.pl记得使用绝对路径,否则找不到
conda activate EDTA
cd EDTA
perl EDTA.pl#测试流程,test文件夹中有我们需要的测试数据,此外这里使用了curatedlib参数,这是一个可选参数,日后若没有已知TE库可不选用该参数。
cd ./EDTA/test
nohup /usr/bin/time -v perl ../EDTA.pl --genome genome.fa --cds genome.cds.fa --curatedlib ../database/rice6.9.5.liban --exclude genome.exclude.bed --overwrite 1 --sensitive 1 --anno 1 --evaluate 1 --threads 10 > EDTA.test &#不出问题的话最后产生的XXX.mod.EDTA.TElib.fa,就是我们需要的最终的TE文库。
三、参数说明
perl EDTA.pl [options]
--genome [File] The genome FASTA file. Required.
--species [Rice|Maize|others] Specify the species for identification of TIR candidates. Default: others
--step [all|filter|final|anno] Specify which steps you want to run EDTA.
all: run the entire pipeline (default)
filter: start from raw TEs to the end.
final: start from filtered TEs to finalizing the run.
anno: perform whole-genome annotation/analysis after TE library construction.
--overwrite [0|1] If previous results are found, decide to overwrite (1, rerun) or not (0, default).
--cds [File] Provide a FASTA file containing the coding sequence (no introns, UTRs, nor TEs) of this genome or its close relative.
--curatedlib [file] Provided a curated library to keep consistant naming and classification for known TEs.
All TEs in this file will be trusted 100%, so please ONLY provide MANUALLY CURATED ones here.
This option is not mandatory. It's totally OK if no file is provided (default).
--sensitive [0|1] Use RepeatModeler to identify remaining TEs (1) or not (0, default).
This step is very slow and MAY help to recover some TEs.
--anno [0|1] Perform (1) or not perform (0, default) whole-genome TE annotation after TE library construction.
--rmout [File] Provide your own homology-based TE annotation instead of using the EDTA library for masking.
File is in RepeatMasker .out format. This file will be merged with the structural-based TE annotation. (--anno 1 required).
Default: use the EDTA library for annotation.
--evaluate [0|1] Evaluate (1) classification consistency of the TE annotation. (--anno 1 required). Default: 0.
This step is slow and does not affect the annotation result.
--exclude [File] Exclude bed format regions from TE annotation. Default: undef. (--anno 1 required).
--u [float] Neutral mutation rate to calculate the age of intact LTR elements.
Intact LTR age is found in this file: *EDTA_raw/LTR/*.pass.list. Default: 1.3e-8 (per bp per year, from rice).
--threads|-t [int] Number of theads to run this script (default: 4)
--help|-h Display this help info#Input说明
Required: 基因组文件[FASTA]。
Optional:
1、Coding sequence of the species or a closely related species [FASTA,帮助清除TE库中的基因序列。
2、Known gene positions of this version of the genome assembly [BED],在此文件中的坐标位置将在TE注释过程中排除,以避免over-masking
3、Curated TE library of the species [FASTA]. This file is trusted 100%. Please make sure it's curated. If you only have a couple of curated sequences, that's fine. It doesn't need to be complete. Providing curated TE sequences, especially for those under annotated TE types (i.e., SINEs and LINEs), will greatly improve the annotation quality.#Output说明详情见官网
#此外,官网还有分步运行perl EDTA_raw.pl,最后使用perl EDTA.pl --overwrite 0自动进行pick up的具体过程。
关于EDTA的更多使用日后再来探索。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号