
SAMtools命令行使用大全
samtools详细的manual page见http://www.htslib.org/doc/
SAM文件格式参数详见http://samtools.github.io/hts-specs/SAMv1.pdf以及http://genome.sph.umich.edu/wiki/SAM
一般使用samtools也就是实现将sam文件转换为bam文件以便于高效处理,再者是samtools stats统计相关的比对信息以及检查sam文件的header信息。这部分内容下文都有涉及。
一、samtools以及sam文件简介
sam文件包括header和alignment两部分:
使用-H参数查看sam/bam文件的header
$ samtools view -H celegans.sam

结合grep查询header指定行
$ samtools view -H celegans.bam | grep "^@RG"

结合head -n参数列出alignment部分第一行
$ samtools view celegans.sam | head -n 1
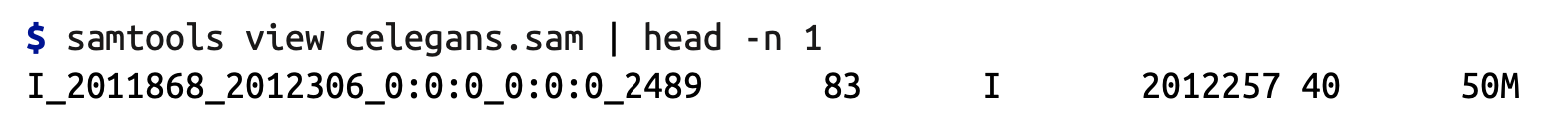
以上命令用于手动检查SAM/BAM文件格式
SAM文件alignment部分,共有11个字段:
使用如下命令查看第一个aligment lines,可使用tr分行显示
$ samtools view celegans.sam | tr '\t' '\n' | head -n 11

依次对应QNAME、FLAG、RNAME、POS、MAPQ、CIGAR、RNEXT/PNEXT、TLEN、SEQ、QUAL(详细信参阅《Bioinformatics Data Skills》第390页)
使用samtools的-b参数完成sam->bam格式转换(sam:纯文本,可读性好;bam:二进制文件,后续处理效率更高)
$ samtools view -b celegans.sam > celegans_copy.bam
使用-h参数将header引入才可以转换回sam文件以供手动检查。
$ samtools view -h celegans.bam > celegans_copy.sam
二、samtools分析指南
SAMtools Sort and Index排序以及建立索引以便更快的比对分析:
排序
$ samtools sort celegans_unsorted.bam celegans_sorted
建立索引
$ samtools index celegans_sorted.bam
现在我们获得了position-sorted和indexed BAM文件
使用samtools查阅1号染色体对应位置上的alignments信息,显示前三行
$ samtools view celegans_sorted.bam 1:215906469-215906652 | head -n 3
或者使用-b参数将其保存为新的BAM文件写入磁盘
$ samtools view -b celegans_sorted.bam 1:215906469-215906652 > specific_celegans_alns.bam
使用-L参数从bed文件中提取具体区域
$ samtools view -L specific_exons.bed celegans_sorted.bam
更多信息使用samtools view查阅Usage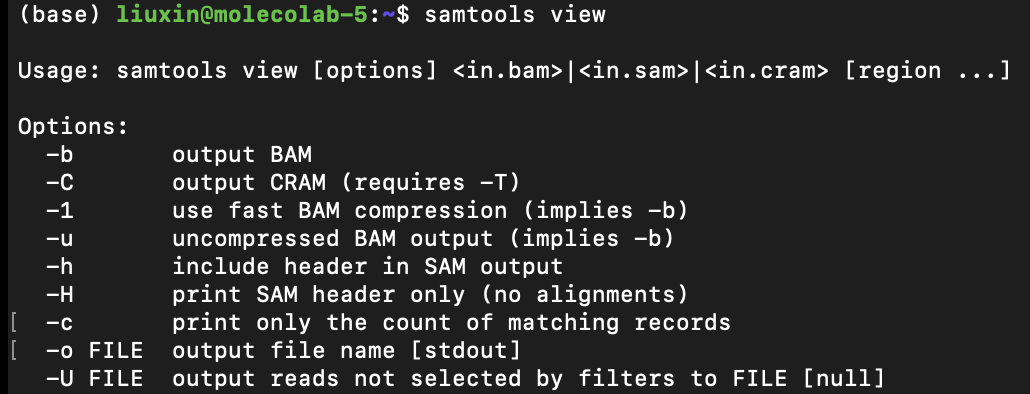
使用samtools flags查阅flags标志位对应信息以便后续过滤分析(如其中flag值0x4代表未比对上的)
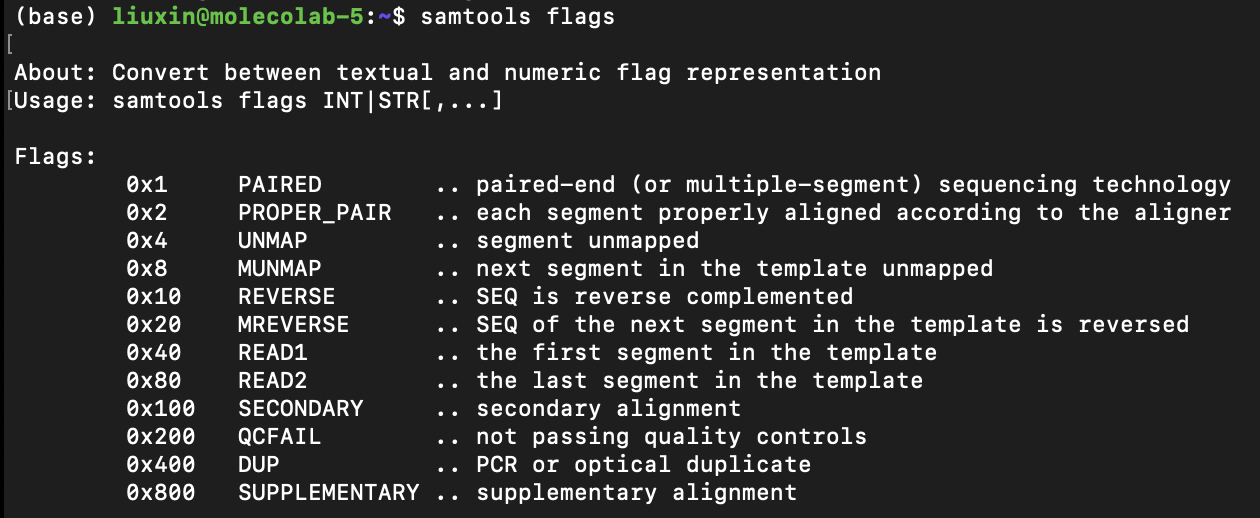
也可以按位组合,例如要过滤未通过质量控制以及未比对上的reads时,使用samtools flags unmap,qcfail查阅value

随后可使用如下命令过滤未通过质量控制以及未比对上的reads,并存盘为新的BAM文件
$ samtools view -b -F 516 celegans_sorted.bam > celegans_Filtering.bam
-F以及-f参数也可以组合使用
使用命令行可视化工具samtools tview可视化部分Alignments,首先查阅Usage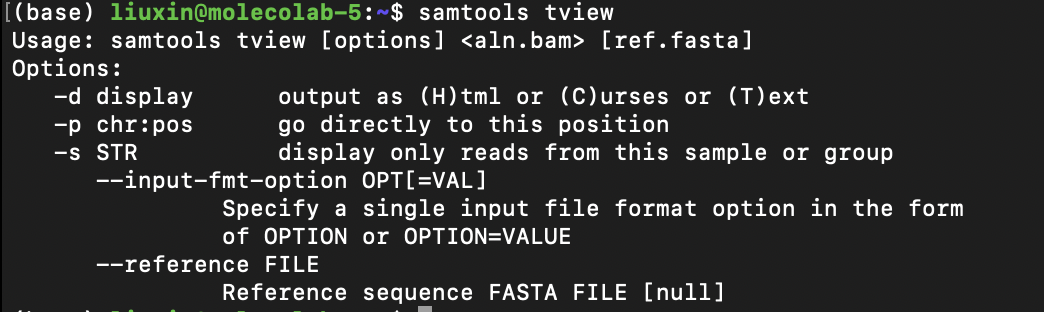
$ samtools tview -p 1:999-1999 <aln.bam> [ref.fasta]
进一步可视化可以使用geneious或者IGV(详见《Bioinformatics Data Skill》第402页)
三、samtools-based variant calling pipelines 结合bcftools
第一步mpileup搭建pileup格式,包含比对数据每个碱基的总结信息,是samtools call variant的第一步
$ samtools mpileup --no-BAQ --region 1:215906528-215906567 --fasta-ref human_g1k_v37.fasta NA12891_CEU_sample.bam
第二步samtools+bcftools两步call variant
$ samtools mpileup -v --no-BAQ --region 1:215906528-215906567 --fasta-ref human_g1k_v37.fasta NA12891_CEU_sample.bam > NA12891_CEU_sample.vcf.gz
$ bcftools call -v -m NA12891_CEU_sample.vcf.gz > NA12891_CEU_sample_calls.vcf.gz
参考自《Bioinformatics Data Skills》

 浙公网安备 33010602011771号
浙公网安备 33010602011771号