
samtools flagstat参数对比对的bam文件进行统计 解读
001、命令
samtools flagstat sample_name.sorted.bam > sample_name.flagstat.txt ## 基本命令

a、生成的文件是一个包含16行的文本文件:
002、
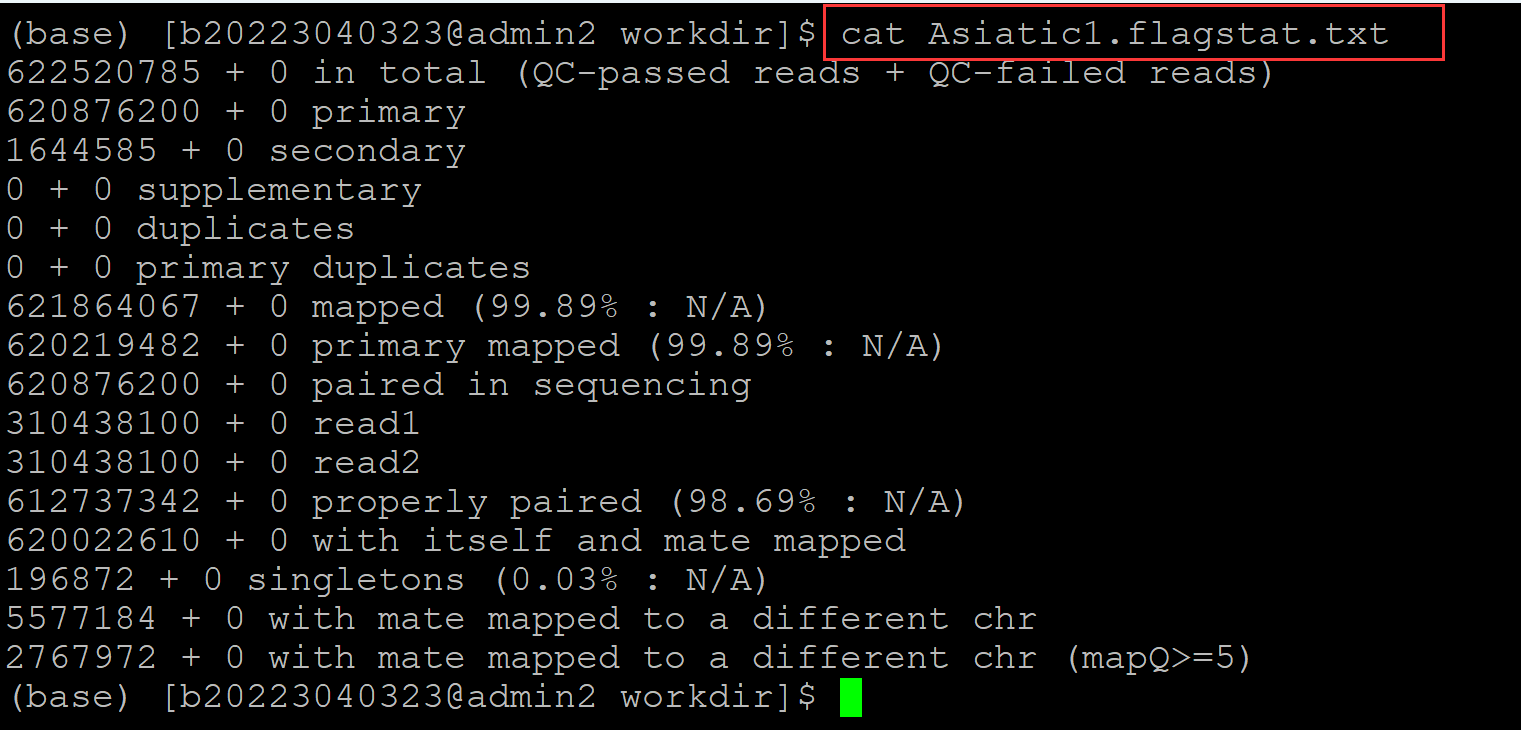
(base) [b20223040323@admin2 workdir]$ cat Asiatic1.flagstat.txt ## 查看统计结果
622520785 + 0 in total (QC-passed reads + QC-failed reads) ## 不是总的reads , 为什么会出现奇数呢,是因为第二行和第三行之和,即原始的reads数目加上比对到多个位置的reads数目,即 620876200 + 1644585;为什么统计这个数字??
620876200 + 0 primary ## 这个地方是总的read数目!!!! fastq1 + fsatq2 的reads总和
1644585 + 0 secondary ## 出现比对到参考基因组多个位置的reads数目
0 + 0 supplementary ## 可能存在嵌合的reads数目,???? 嵌合是什么意思??
0 + 0 duplicates ## PCR重复的reads数目
0 + 0 primary duplicates ## ??为社么又出现一个重复的??
621864067 + 0 mapped (99.89% : N/A) ## 正常比对的reads + 比对到参考基因组多个位置的reads之和,这里 644585 + 196872 + 620022610; 比对率再除以 第一行统计到的reads数目,这个值有何意义??
1644620219482 + 0 primary mapped (99.89% : N/A) ## 原始的比对率,双端比对到参考基因组的reads数,单端比对到参考基因组的reads数,这里 196872 + 620022610
620876200 + 0 paired in sequencing ## 双端测序的reads数目, 难道还会有其他的??
310438100 + 0 read1 ## read1的数目
310438100 + 0 read2 ## read2的数目
612737342 + 0 properly paired (98.69% : N/A) ## 完美比对的reads数,两个reads比对到同一个DNA片段,同时距离符合设置的阈值, 这里 612737342 / 620876200
620022610 + 0 with itself and mate mapped
196872 + 0 singletons (0.03% : N/A) ## 双端reads中,其中一端比对上,另一端没有比对上的read 数目
5577184 + 0 with mate mapped to a different chr ## 比对到两条不同序列的reads数目
2767972 + 0 with mate mapped to a different chr (mapQ>=5)
。
如果有些结果全部为0,可以检查一下是否对这些参数进行标记。比如标记重复MarkDuplicates。(https://www.jianshu.com/p/34e3e3853fd5)
这个地方是否提示,在利用samtools进行基本统计的时候,是否必须使用标记过后的bam文件进行统计。
003、
参考:
001、https://www.jianshu.com/p/34e3e3853fd5
002、https://www.jianshu.com/p/ccc59b459d4a

 浙公网安备 33010602011771号
浙公网安备 33010602011771号