
Linux 中 2>&1 解释
在Linux系统中:
0 表示标准输入;
1表示标准输出;
2表示标准错误输出;
2>&1 表示将标准错误输出重定向到标准输入;
举一个例子:
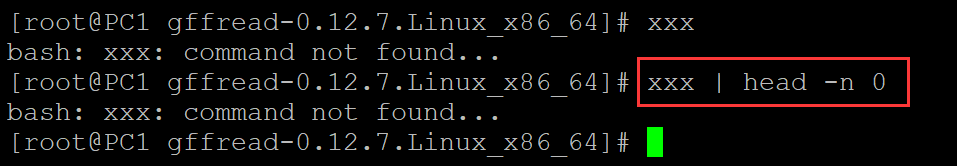
a、不将标准错误输出 重定向到标准输入中。
[root@PC1 gffread-0.12.7.Linux_x86_64]# xxx ## 在终端随机输入一个命令,是一个错误输出 bash: xxx: command not found... [root@PC1 gffread-0.12.7.Linux_x86_64]# xxx | head -n 0 ## 结合管道,表明左侧的命令没有进入管道,也就是说标准错误输出直接输出到终端了。 bash: xxx: command not found...
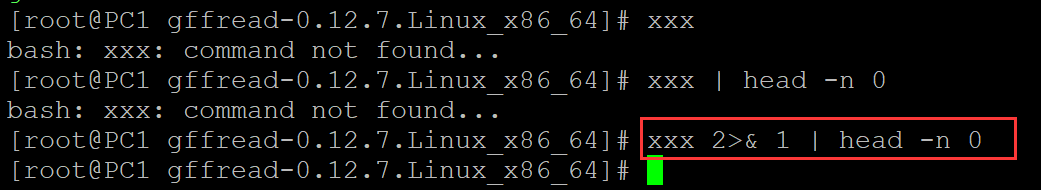
b、将标准错误输出重定向到标准输入中。
[root@PC1 gffread-0.12.7.Linux_x86_64]# xxx bash: xxx: command not found... [root@PC1 gffread-0.12.7.Linux_x86_64]# xxx | head -n 0 bash: xxx: command not found... [root@PC1 gffread-0.12.7.Linux_x86_64]# xxx 2>& 1 | head -n 0 ## 将标准错误输出重定向到标准输入中
。
举例2:
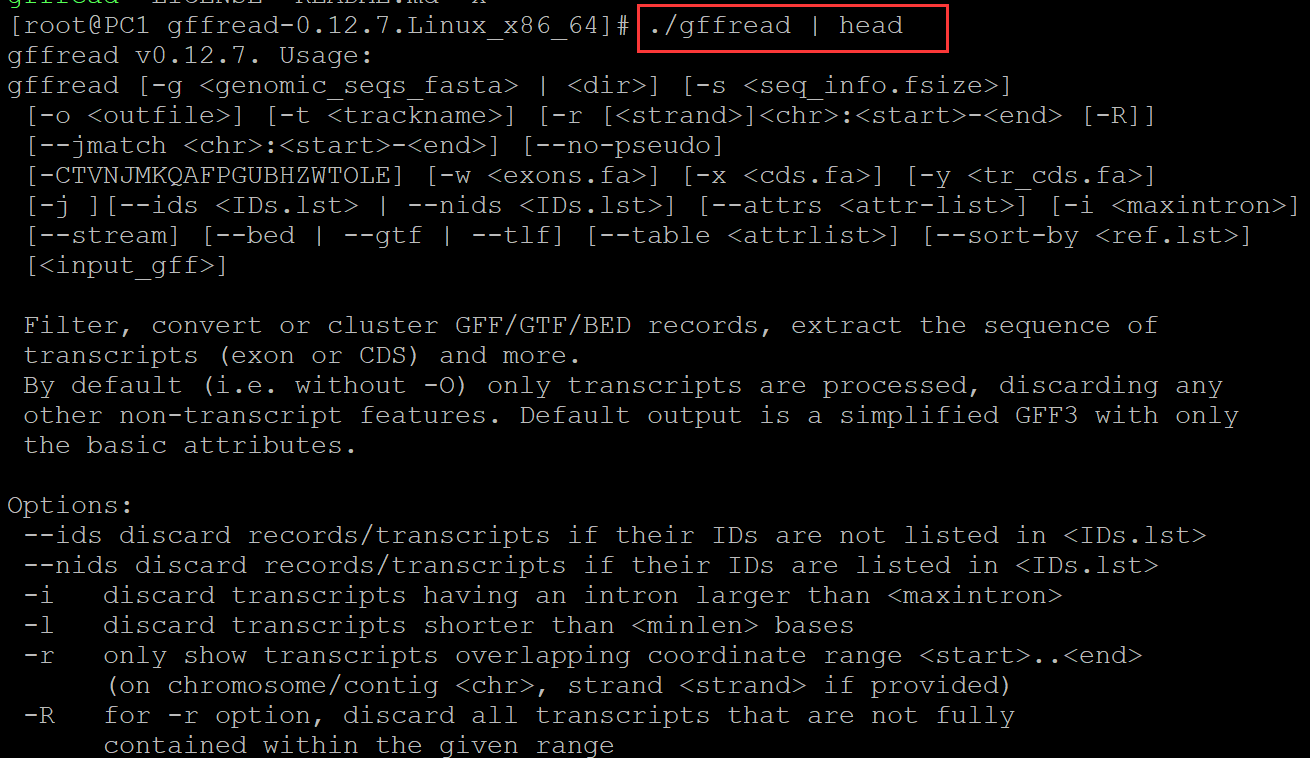
a、

[root@PC1 gffread-0.12.7.Linux_x86_64]# ./gffread | head ## 管道不起作用 gffread v0.12.7. Usage: gffread [-g <genomic_seqs_fasta> | <dir>] [-s <seq_info.fsize>] [-o <outfile>] [-t <trackname>] [-r [<strand>]<chr>:<start>-<end> [-R]] [--jmatch <chr>:<start>-<end>] [--no-pseudo] [-CTVNJMKQAFPGUBHZWTOLE] [-w <exons.fa>] [-x <cds.fa>] [-y <tr_cds.fa>] [-j ][--ids <IDs.lst> | --nids <IDs.lst>] [--attrs <attr-list>] [-i <maxintron>] [--stream] [--bed | --gtf | --tlf] [--table <attrlist>] [--sort-by <ref.lst>] [<input_gff>] Filter, convert or cluster GFF/GTF/BED records, extract the sequence of transcripts (exon or CDS) and more. By default (i.e. without -O) only transcripts are processed, discarding any other non-transcript features. Default output is a simplified GFF3 with only the basic attributes. Options: --ids discard records/transcripts if their IDs are not listed in <IDs.lst> --nids discard records/transcripts if their IDs are listed in <IDs.lst> -i discard transcripts having an intron larger than <maxintron> -l discard transcripts shorter than <minlen> bases -r only show transcripts overlapping coordinate range <start>..<end> (on chromosome/contig <chr>, strand <strand> if provided) -R for -r option, discard all transcripts that are not fully contained within the given range
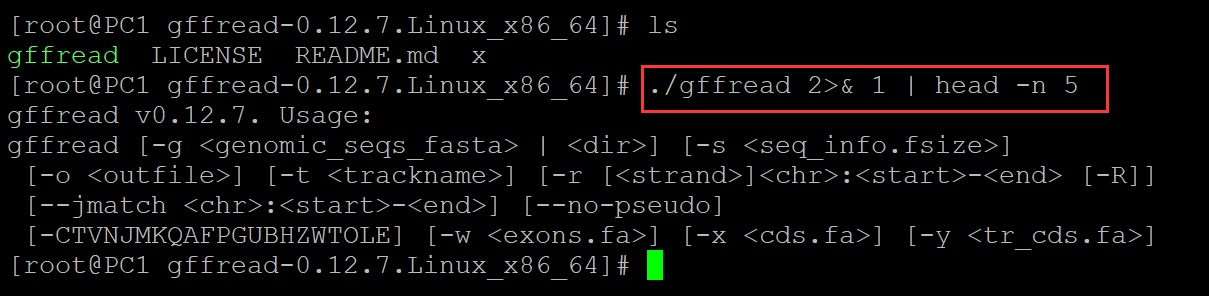
b、管道生效
[root@PC1 gffread-0.12.7.Linux_x86_64]# ls gffread LICENSE README.md x [root@PC1 gffread-0.12.7.Linux_x86_64]# ./gffread 2>& 1 | head -n 5 ## 管道生效,仅仅输出5行 gffread v0.12.7. Usage: gffread [-g <genomic_seqs_fasta> | <dir>] [-s <seq_info.fsize>] [-o <outfile>] [-t <trackname>] [-r [<strand>]<chr>:<start>-<end> [-R]] [--jmatch <chr>:<start>-<end>] [--no-pseudo] [-CTVNJMKQAFPGUBHZWTOLE] [-w <exons.fa>] [-x <cds.fa>] [-y <tr_cds.fa>]
。
分类:
linux shell



【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· 震惊!C++程序真的从main开始吗?99%的程序员都答错了
· 【硬核科普】Trae如何「偷看」你的代码?零基础破解AI编程运行原理
· 单元测试从入门到精通
· 上周热点回顾(3.3-3.9)
· winform 绘制太阳,地球,月球 运作规律
2023-05-08 linux 中xargs -i 和-I在使用上的区别
2023-05-08 linux 中 xargs 中的 -i选项
2022-05-08 R语言中实现第一列转化为行名、第一行转化为列名
2022-05-08 R语言中利用readxl包读取excel数据
2022-05-08 R语言中获取变量占据内存的大小object.size函数
2022-05-08 R语言实现数据的标准化
2022-05-08 R语言中scale函数的用法