
samtools flagstat 参数对bam文件统计结果解读
001、使用命令 及生成结果
samtools flagstat -@ 56 ERR3143219.sort.bam > flagstat.txt ## samtools flagstat统计
002、输出结果解读

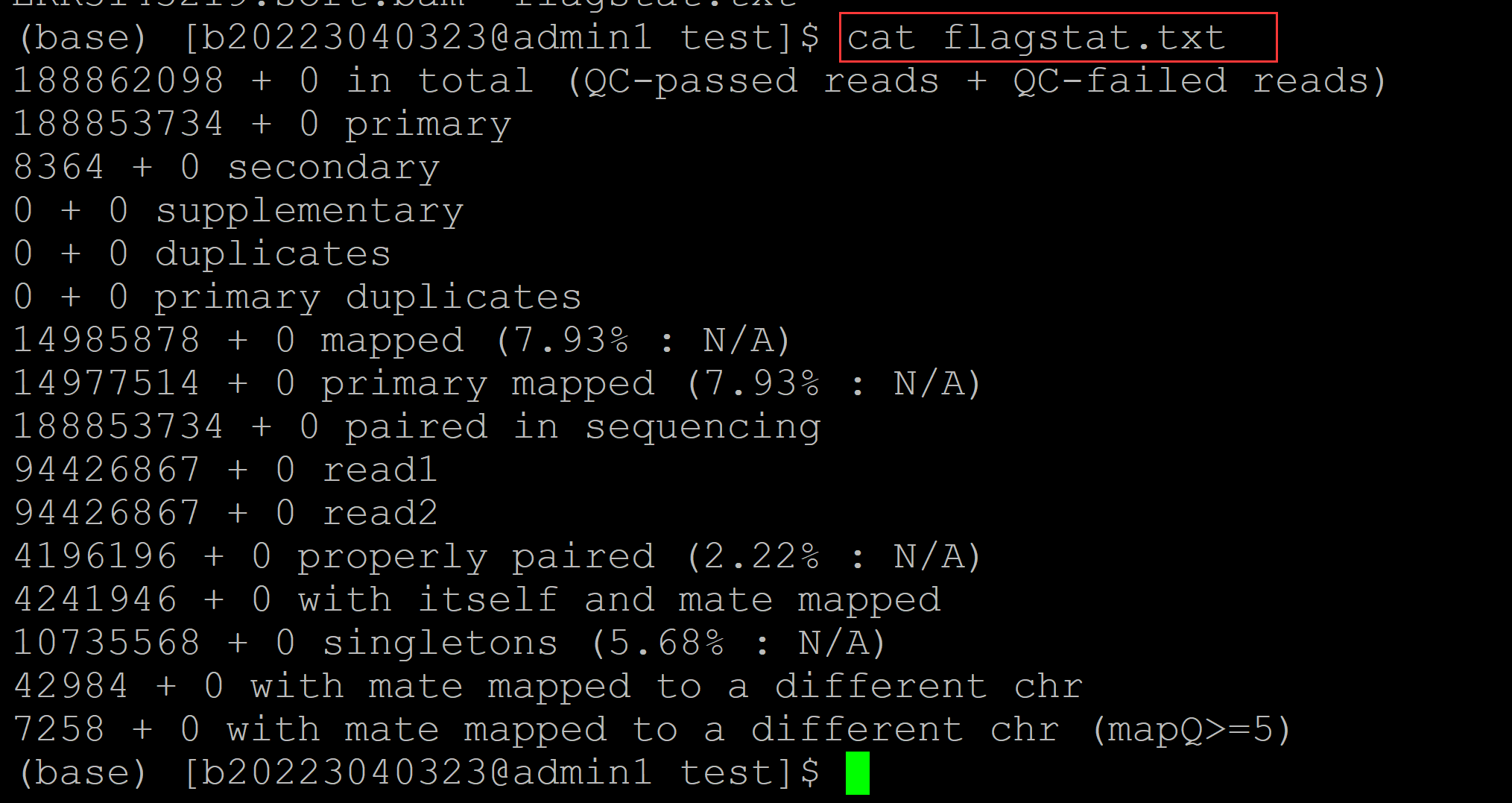
(base) [b20223040323@admin1 test]$ cat flagstat.txt ## 会多于原始的fastq中的reads数目,因为存在比对到参考基因组多个位置的情况 188862098 + 0 in total (QC-passed reads + QC-failed reads) ## 比对到参考基因组总的read数目,包括原始的fastq的总的reads数和和比对到参考基因组多个位置reads的计数 188853734 + 0 primary ## 原始的fastq中的read数目 8364 + 0 secondary ## 比对到参考基因组多个位置的reads数目 0 + 0 supplementary ## 可能存在嵌合的reads数目 0 + 0 duplicates ## 重复的reads数目 0 + 0 primary duplicates 14985878 + 0 mapped (7.93% : N/A) ## 比对到参考基因组的reads计数(包括比对到参考基因组多个位置的reads计数)占reads总数的比例 14977514 + 0 primary mapped (7.93% : N/A) ## 原始reads比对到参考基因组的数目占原始reads的比例 188853734 + 0 paired in sequencing ## 双端测序的数目(实际就是reads数) 94426867 + 0 read1 ## read1的数目 94426867 + 0 read2 ## read2的数目 4196196 + 0 properly paired (2.22% : N/A) ## 完美比对的reads数目(双端reads比对到同一条序列,且根据比对结果推断的插入片段大小符合设置的阈值) 4241946 + 0 with itself and mate mapped ## 双端reads均比对到参考基因组的数目 10735568 + 0 singletons (5.68% : N/A) ## 双端reads,一端比对上, 另一端没有比对上的reads数目 42984 + 0 with mate mapped to a different chr ## 比对到两条序列的reads数目 7258 + 0 with mate mapped to a different chr (mapQ>=5) ## 且mapQ >= 5的数目
。
参考:
01、https://wenku.baidu.com/view/94ef844924d3240c844769eae009581b6bd9bd96.html?_wkts_=1699923563989&bdQuery=samtools+flagstat+%E5%8F%82%E6%95%B0+%E7%BB%93%E6%9E%9C%E8%A7%A3%E8%AF%BB



【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· 震惊!C++程序真的从main开始吗?99%的程序员都答错了
· 【硬核科普】Trae如何「偷看」你的代码?零基础破解AI编程运行原理
· 单元测试从入门到精通
· 上周热点回顾(3.3-3.9)
· winform 绘制太阳,地球,月球 运作规律
2022-11-14 python 中 #-*-coding: UTF-8 -*-的作用
2022-11-14 python 中实现在命令行中传递参数
2020-11-14 ansible