
seqkit软件根据染色体名称从fasta文件中批量提取数据
001、

[root@pc1 test1]# ls a.fa chr.list [root@pc1 test1]# cat a.fa ## 测试fasta >chr1 tttcccggg >chr2 tttggg ccc >chr3 cccttt >chr4 aaaaattt [root@pc1 test1]# cat chr.list ## 染色体列表 chr2 chr4 [root@pc1 test1]# seqkit -w 8 grep -f chr.list a.fa > result.fa ## -w指定每行输出的碱基数目 [INFO] 2 patterns loaded from file [root@pc1 test1]# cat result.fa ## 结果文件 >chr2 tttgggcc c >chr4 aaaaattt
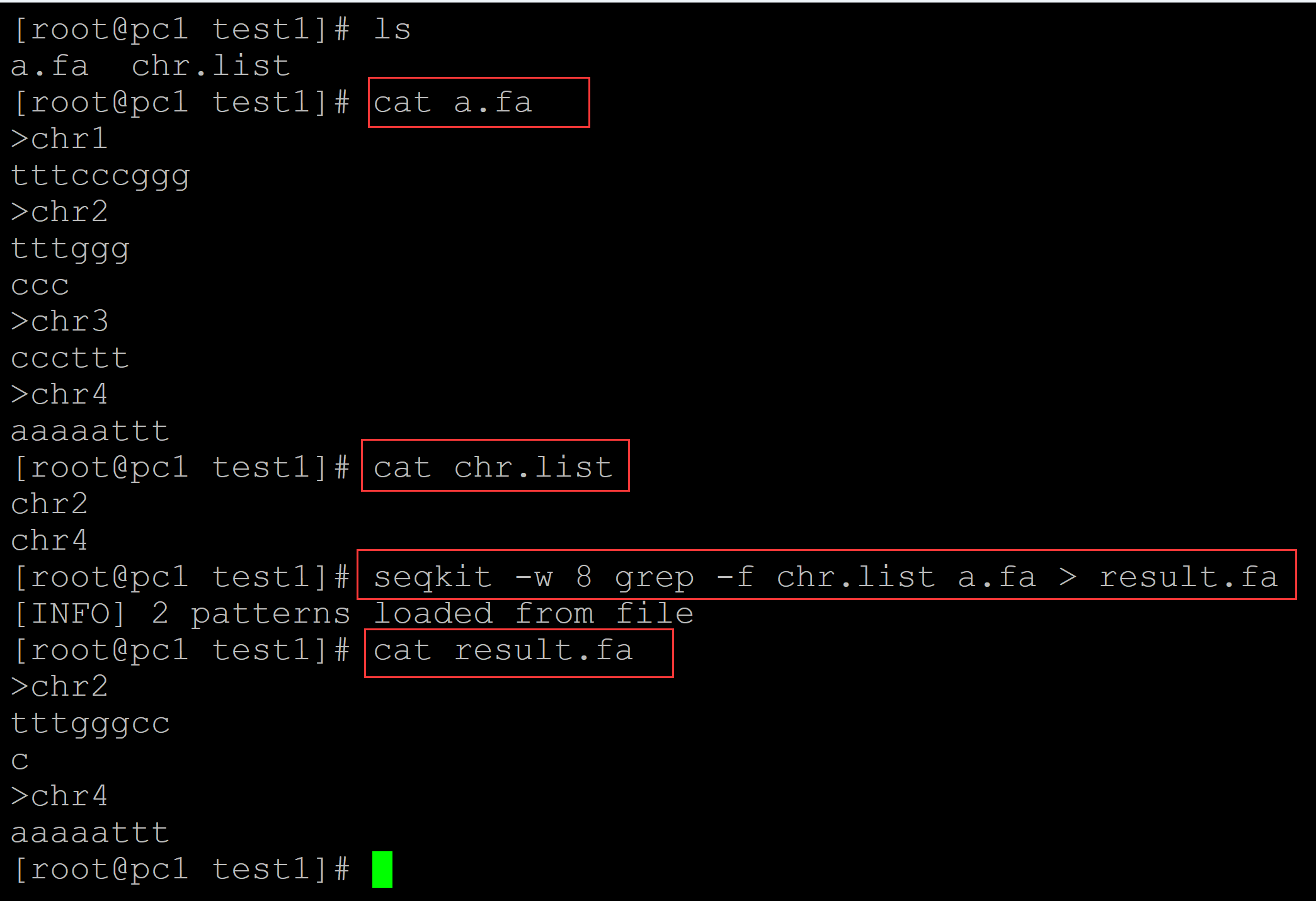
。



【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· 震惊!C++程序真的从main开始吗?99%的程序员都答错了
· 【硬核科普】Trae如何「偷看」你的代码?零基础破解AI编程运行原理
· 单元测试从入门到精通
· 上周热点回顾(3.3-3.9)
· winform 绘制太阳,地球,月球 运作规律
2020-10-13 linux文件测试语句
2020-10-13 linux 系统中while循环示例
2020-10-13 linux系统中$RANDOM命令
2020-10-13 linux系统中let命令
2020-10-13 linux系统中expr命令
2020-10-13 linux系统中 read命令