
seqkit 软件的安装及使用
001、安装
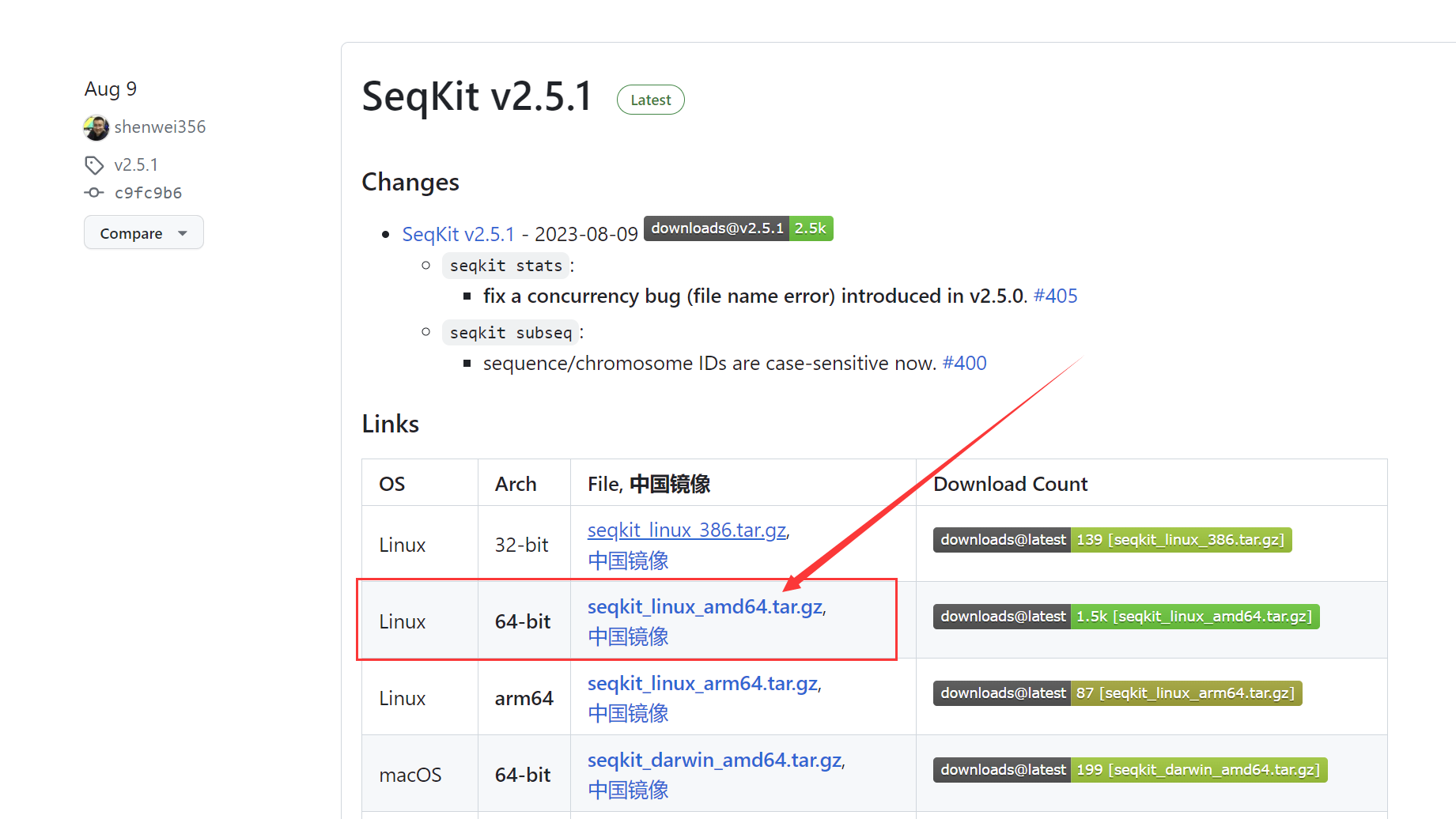
下载最新版:
官网:https://github.com/shenwei356/seqkit/releases
002、解压、调用测试

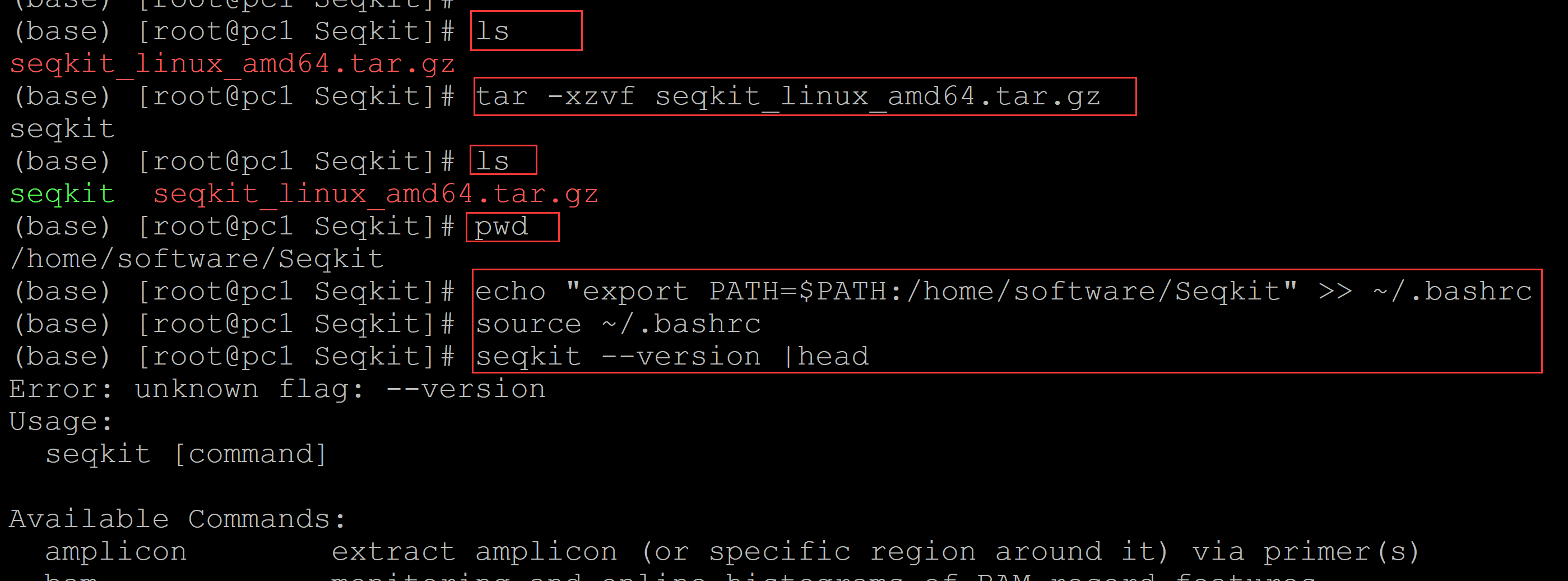
(base) [root@pc1 Seqkit]# ls ## 安装包 seqkit_linux_amd64.tar.gz (base) [root@pc1 Seqkit]# tar -xzvf seqkit_linux_amd64.tar.gz ## 解压 seqkit (base) [root@pc1 Seqkit]# ls seqkit seqkit_linux_amd64.tar.gz (base) [root@pc1 Seqkit]# pwd /home/software/Seqkit (base) [root@pc1 Seqkit]# echo "export PATH=$PATH:/home/software/Seqkit" >> ~/.bashrc ## 添加环境变量 (base) [root@pc1 Seqkit]# source ~/.bashrc ## 加载环境变量 (base) [root@pc1 Seqkit]# seqkit --version |head ## 调用测试 Error: unknown flag: --version Usage: seqkit [command] Available Commands: amplicon extract amplicon (or specific region around it) via primer(s) bam monitoring and online histograms of BAM record features
003、 -p参数取互补序列
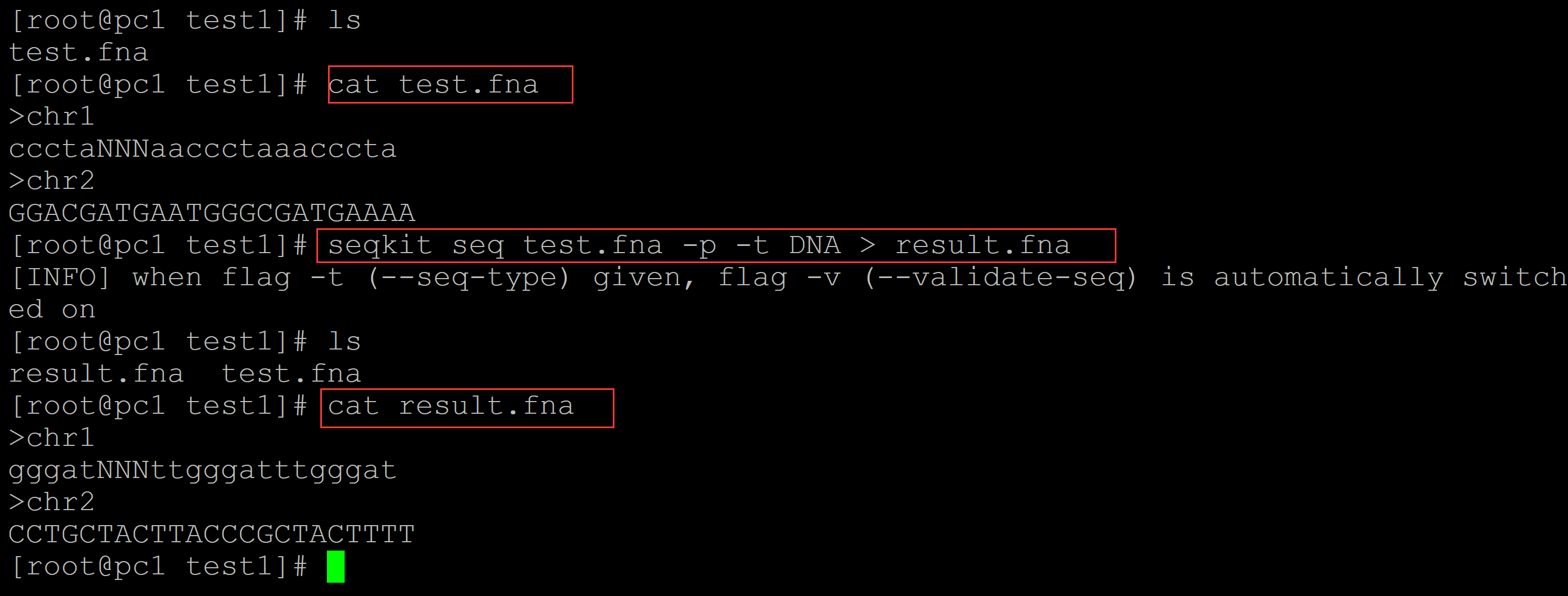
[root@pc1 test1]# ls test.fna [root@pc1 test1]# cat test.fna ## 测试fasta序列 >chr1 ccctaNNNaaccctaaacccta >chr2 GGACGATGAATGGGCGATGAAAA [root@pc1 test1]# seqkit seq test.fna -p -t DNA > result.fna ## 取互补序列 [INFO] when flag -t (--seq-type) given, flag -v (--validate-seq) is automatically switched on [root@pc1 test1]# ls result.fna test.fna [root@pc1 test1]# cat result.fna ## 互补序列结果 >chr1 gggatNNNttgggatttgggat >chr2 CCTGCTACTTACCCGCTACTTTT
004、-r 取反向序列
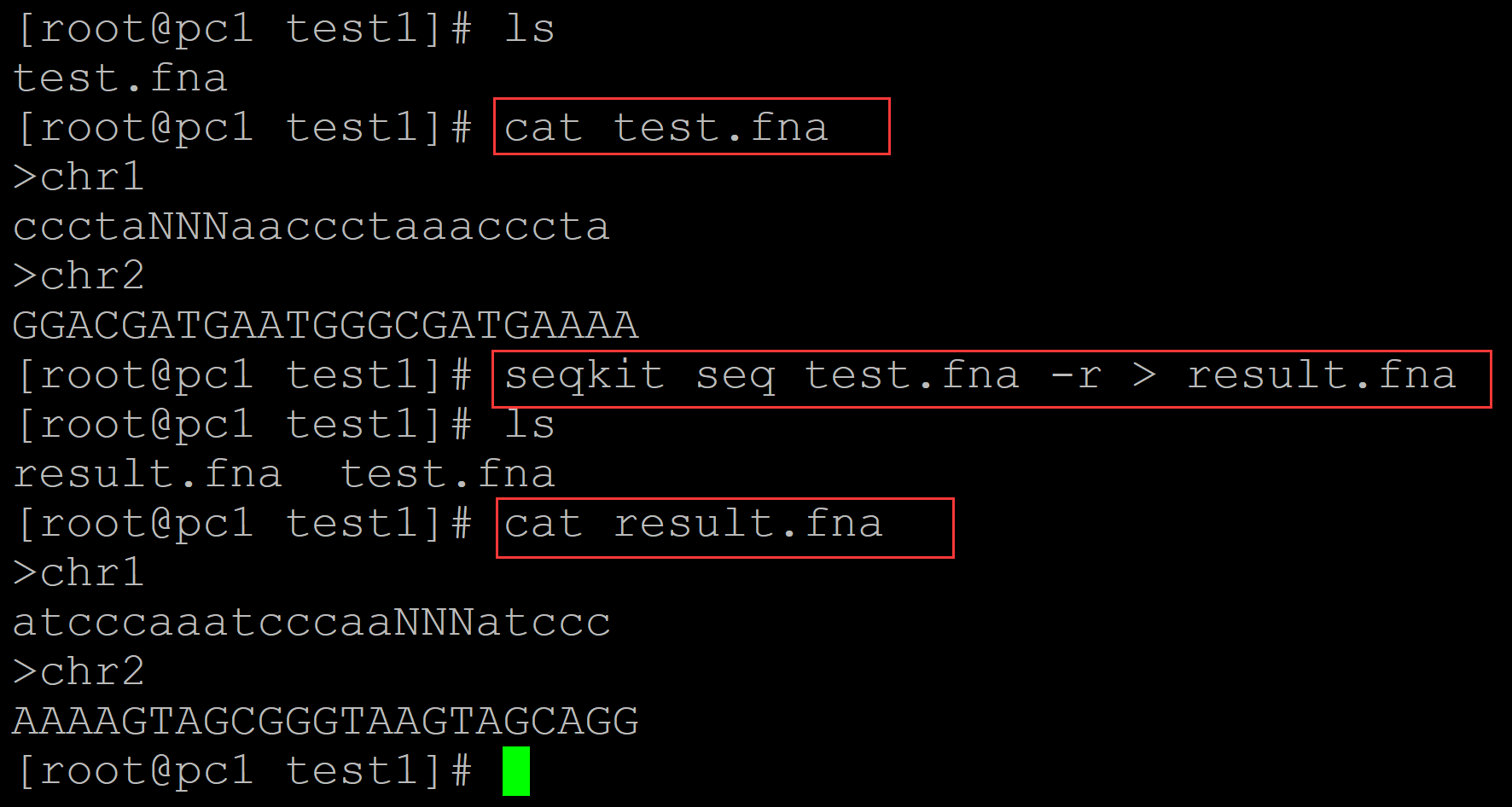
[root@pc1 test1]# ls test.fna [root@pc1 test1]# cat test.fna ## 测试fasta >chr1 ccctaNNNaaccctaaacccta >chr2 GGACGATGAATGGGCGATGAAAA [root@pc1 test1]# seqkit seq test.fna -r > result.fna ## 取反向序列 [root@pc1 test1]# ls result.fna test.fna [root@pc1 test1]# cat result.fna ## 结果文件 >chr1 atcccaaatcccaaNNNatccc >chr2 AAAAGTAGCGGGTAAGTAGCAGG
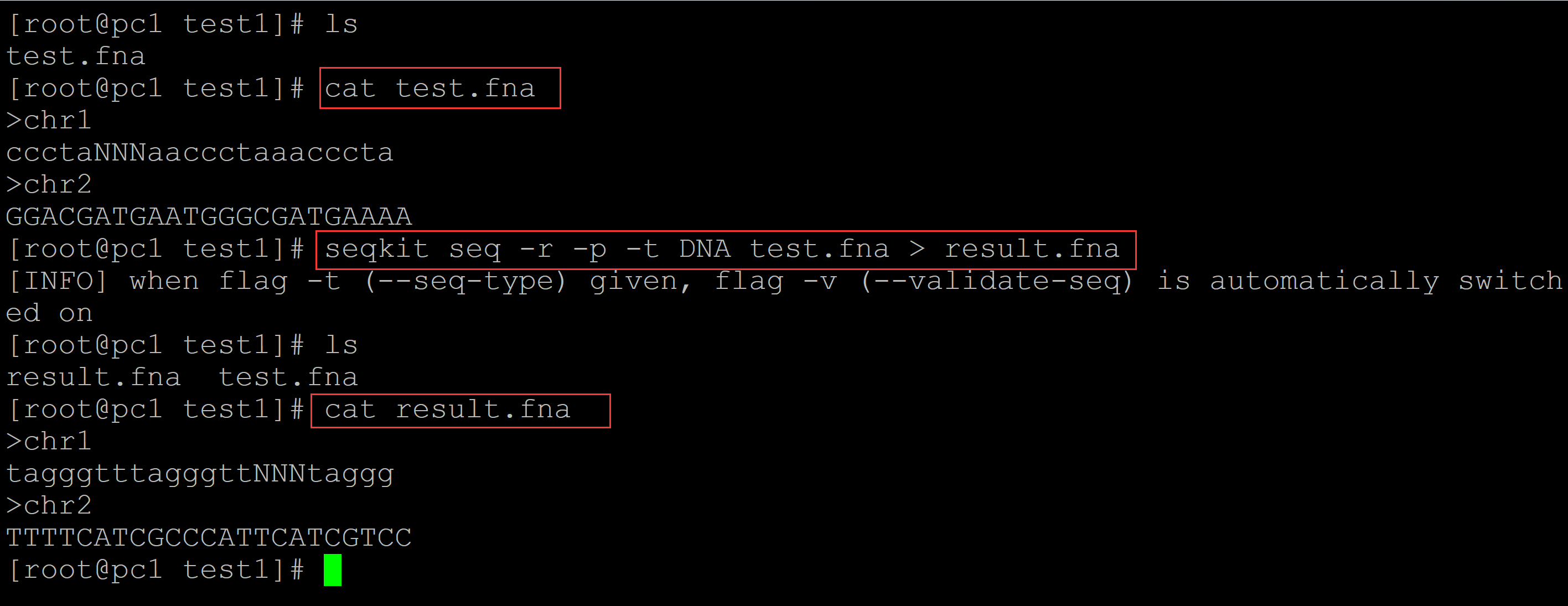
005、-r -p取反向互补序列
[root@pc1 test1]# ls test.fna [root@pc1 test1]# cat test.fna ## 测试数据 >chr1 ccctaNNNaaccctaaacccta >chr2 GGACGATGAATGGGCGATGAAAA [root@pc1 test1]# seqkit seq -r -p -t DNA test.fna > result.fna ## 取反向互补序列 [INFO] when flag -t (--seq-type) given, flag -v (--validate-seq) is automatically switched on [root@pc1 test1]# ls result.fna test.fna [root@pc1 test1]# cat result.fna ## 结果文件 >chr1 tagggtttagggttNNNtaggg >chr2 TTTTCATCGCCCATTCATCGTCC
006、DNA转换为RNA序列
[root@pc1 test1]# ls test.fna [root@pc1 test1]# cat test.fna ## 测试fasta >chr1 ccctaNNNaaccctaaacccta >chr2 GGACGATGAATGGGCGATGAAAA [root@pc1 test1]# seqkit seq test.fna --dna2rna > result.fna ## DNA序列转换为RNA序列
[root@pc1 test1]# ls result.fna test.fna [root@pc1 test1]# cat result.fna ## 转换结果 >chr1 cccuaNNNaacccuaaacccua >chr2 GGACGAUGAAUGGGCGAUGAAAA

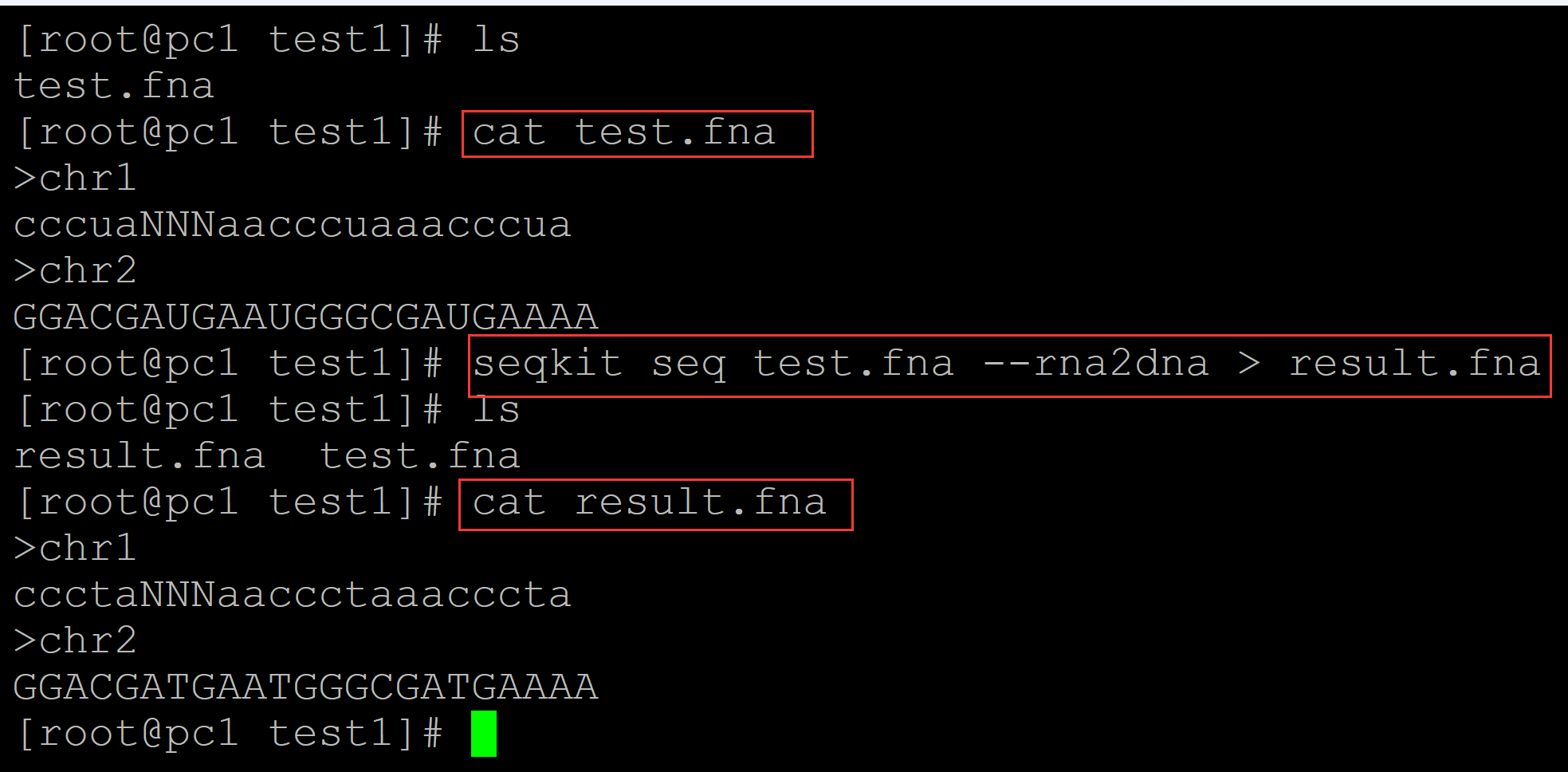
007、RNA序列转换为DNA序列
[root@pc1 test1]# ls test.fna [root@pc1 test1]# cat test.fna ## 测试数据 >chr1 cccuaNNNaacccuaaacccua >chr2 GGACGAUGAAUGGGCGAUGAAAA [root@pc1 test1]# seqkit seq test.fna --rna2dna > result.fna ## rna转换为dna [root@pc1 test1]# ls result.fna test.fna [root@pc1 test1]# cat result.fna ## 结果文件 >chr1 ccctaNNNaaccctaaacccta >chr2 GGACGATGAATGGGCGATGAAAA
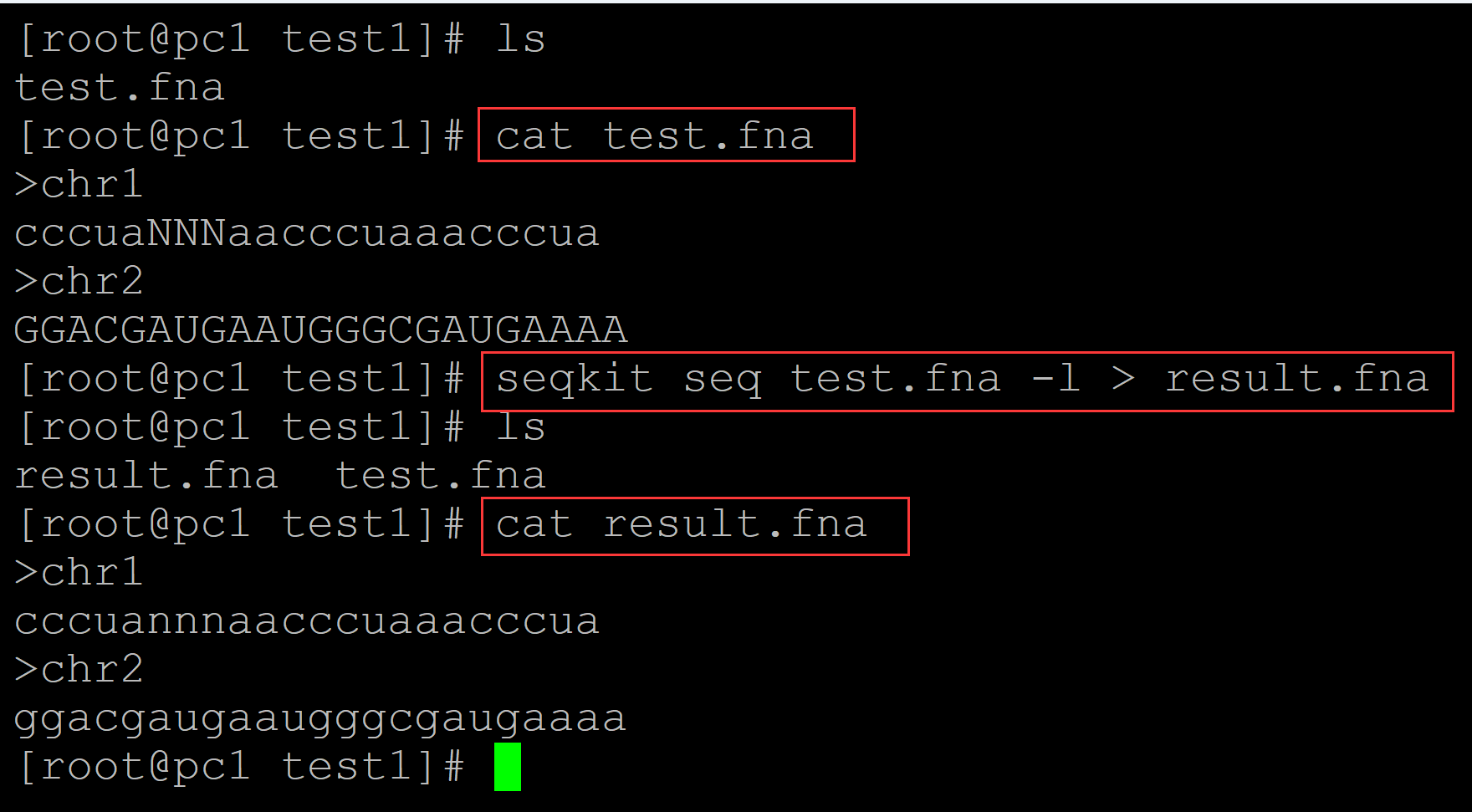
008、以小写字母的形式输出
[root@pc1 test1]# ls test.fna [root@pc1 test1]# cat test.fna ## 测试数据 >chr1 cccuaNNNaacccuaaacccua >chr2 GGACGAUGAAUGGGCGAUGAAAA [root@pc1 test1]# seqkit seq test.fna -l > result.fna ## 小写字母形式输出 [root@pc1 test1]# ls result.fna test.fna [root@pc1 test1]# cat result.fna ## 结果文件 >chr1 cccuannnaacccuaaacccua >chr2 ggacgaugaaugggcgaugaaaa
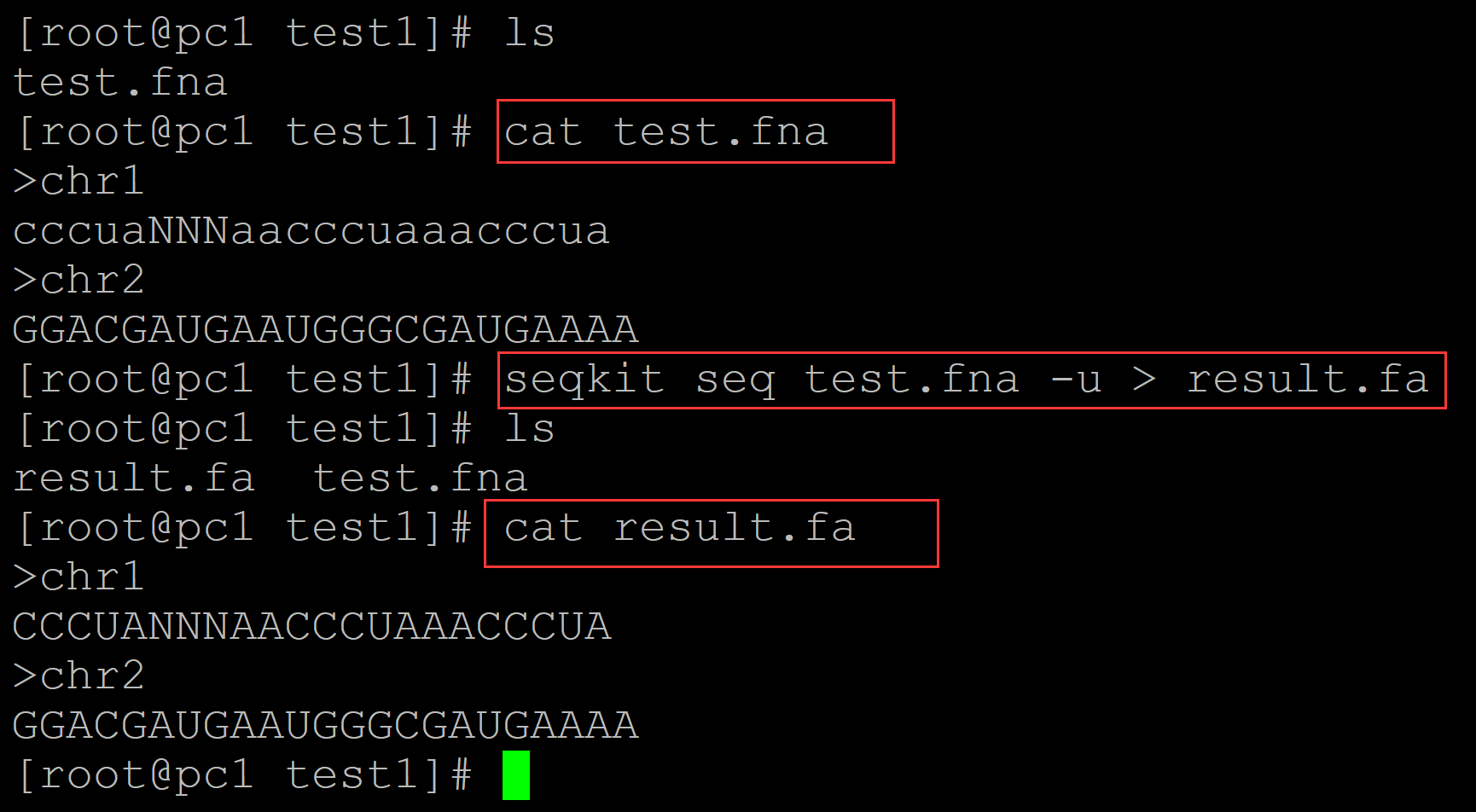
009、序列以大写字母的形式输出
[root@pc1 test1]# ls test.fna [root@pc1 test1]# cat test.fna ## 测试数据 >chr1 cccuaNNNaacccuaaacccua >chr2 GGACGAUGAAUGGGCGAUGAAAA [root@pc1 test1]# seqkit seq test.fna -u > result.fa ## 转换为大写字母 [root@pc1 test1]# ls result.fa test.fna [root@pc1 test1]# cat result.fa ## 结果文件 >chr1 CCCUANNNAACCCUAAACCCUA >chr2 GGACGAUGAAUGGGCGAUGAAAA
010、每行的碱基以指定的数目输出
[root@pc1 test1]# ls test.fna [root@pc1 test1]# cat test.fna ## 测试数据 >chr1 cccuaNNNaacccuaaacccua >chr2 GGACGAUGAAUGGGCGAUGAAAA [root@pc1 test1]# seqkit seq test.fna -w 10 > result.fa ## 每行转换为10个碱基 [root@pc1 test1]# ls result.fa test.fna [root@pc1 test1]# cat result.fa ## 结果文件 >chr1 cccuaNNNaa cccuaaaccc ua >chr2 GGACGAUGAA UGGGCGAUGA AAA

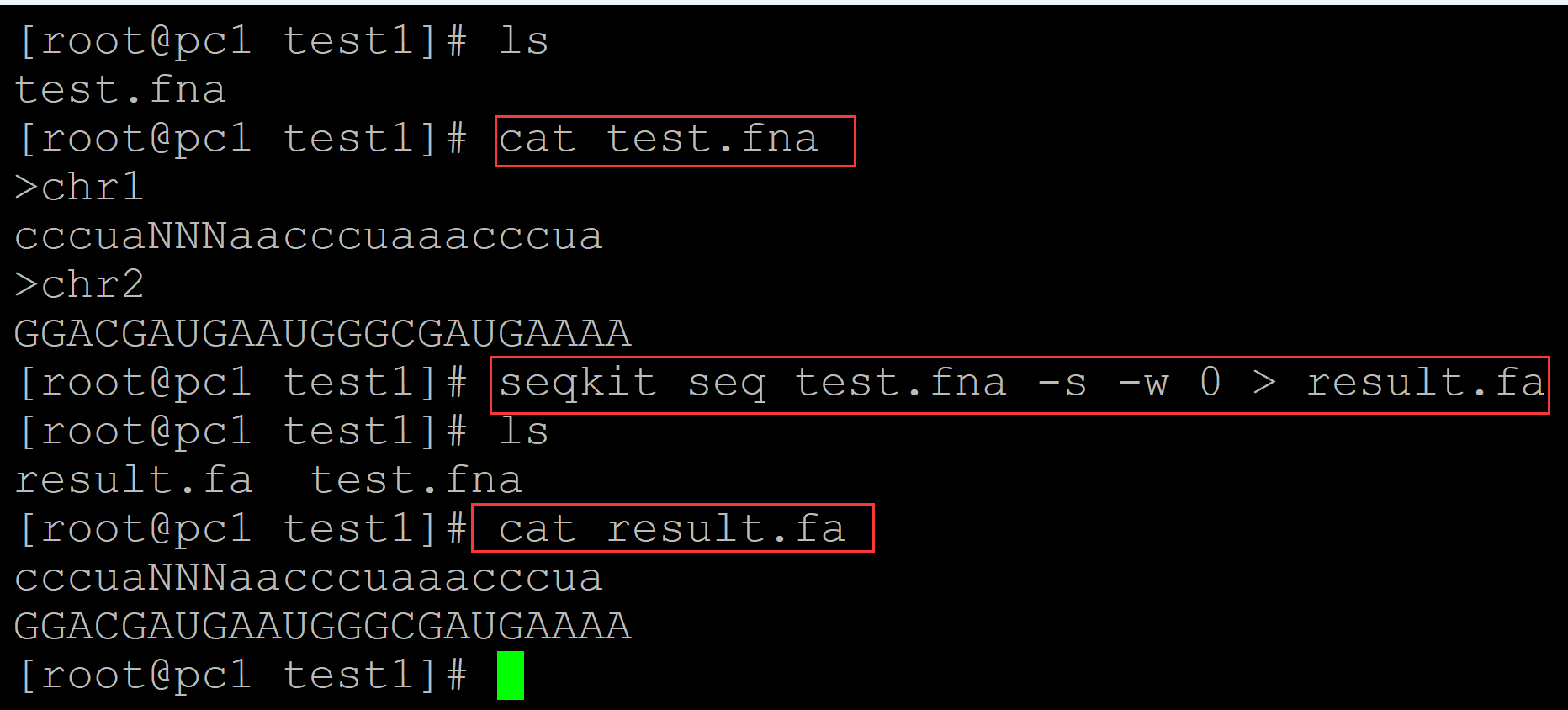
011、仅提取序列文件
[root@pc1 test1]# ls test.fna [root@pc1 test1]# cat test.fna ## 测试fasta >chr1 cccuaNNNaacccuaaacccua >chr2 GGACGAUGAAUGGGCGAUGAAAA [root@pc1 test1]# seqkit seq test.fna -s -w 0 > result.fa ## 仅提取序列文件 [root@pc1 test1]# ls result.fa test.fna [root@pc1 test1]# cat result.fa ## 结果文件 cccuaNNNaacccuaaacccua GGACGAUGAAUGGGCGAUGAAAA
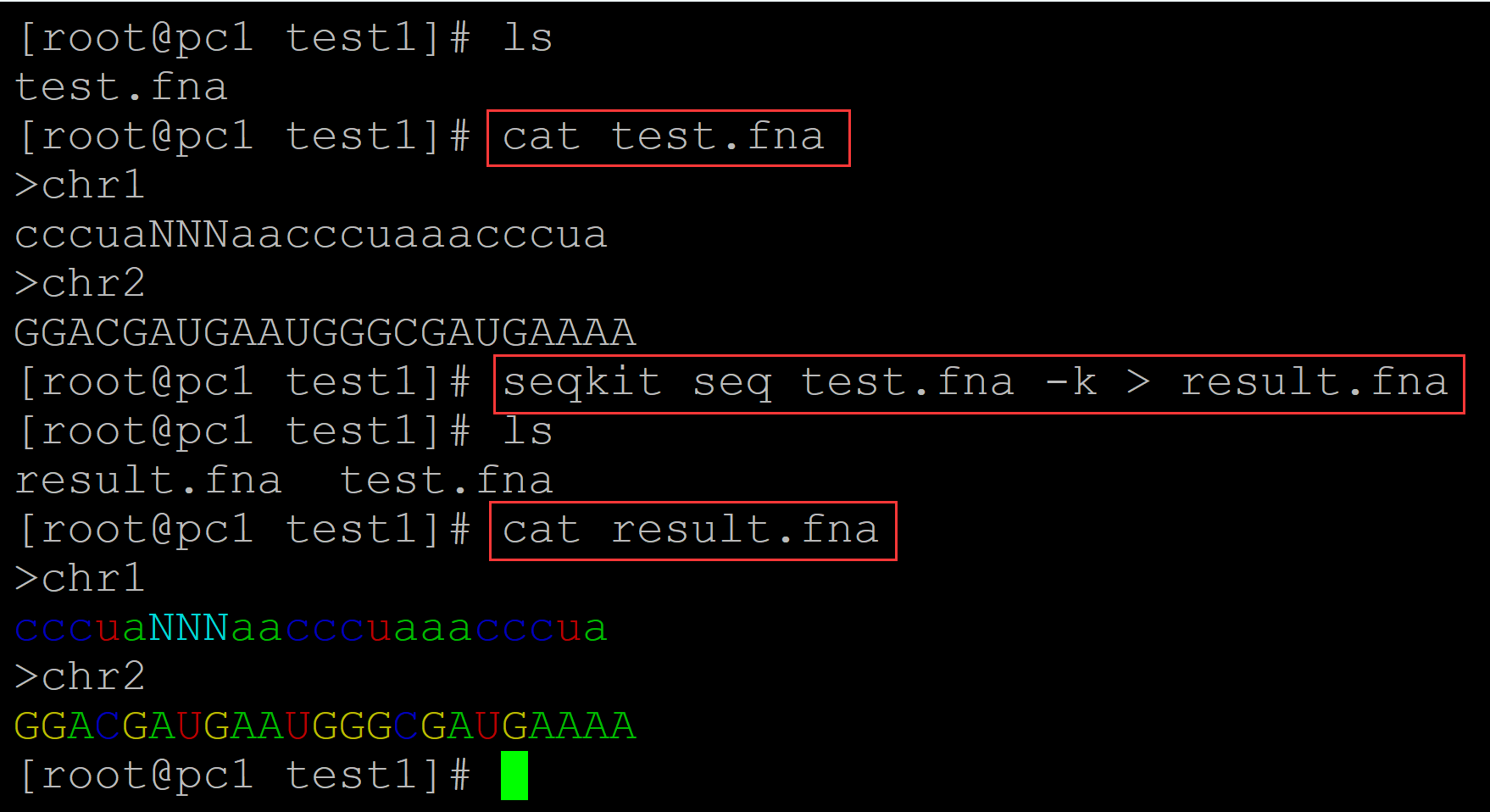
012、输出颜色
[root@pc1 test1]# ls test.fna [root@pc1 test1]# cat test.fna ## 测试文件 >chr1 cccuaNNNaacccuaaacccua >chr2 GGACGAUGAAUGGGCGAUGAAAA [root@pc1 test1]# seqkit seq test.fna -k > result.fna ## 输出颜色 [root@pc1 test1]# ls result.fna test.fna [root@pc1 test1]# cat result.fna ## 结果 >chr1 cccuaNNNaacccuaaacccua >chr2 GGACGAUGAAUGGGCGAUGAAAA
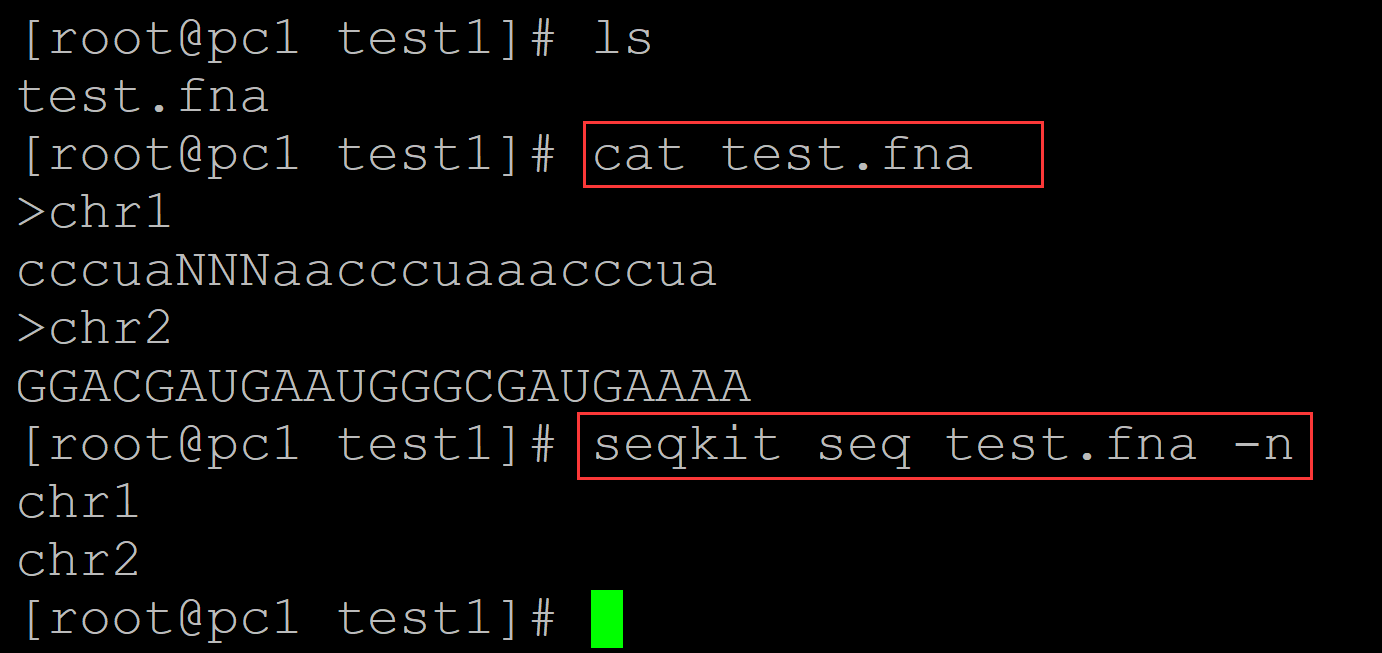
013、输出序列全名
[root@pc1 test1]# ls test.fna [root@pc1 test1]# cat test.fna ## 测试文件 >chr1 cccuaNNNaacccuaaacccua >chr2 GGACGAUGAAUGGGCGAUGAAAA [root@pc1 test1]# seqkit seq test.fna -n ## 输出全名 chr1 chr2
014、
[root@pc1 test1]# ls test.fna [root@pc1 test1]# cat test.fna >chr1 cccuaNNNaacccuaaacccua >chr2 GGACGAUGAAUGGGCGAUGAAAA [root@pc1 test1]# seqkit seq test.fna -i -n chr1 chr2

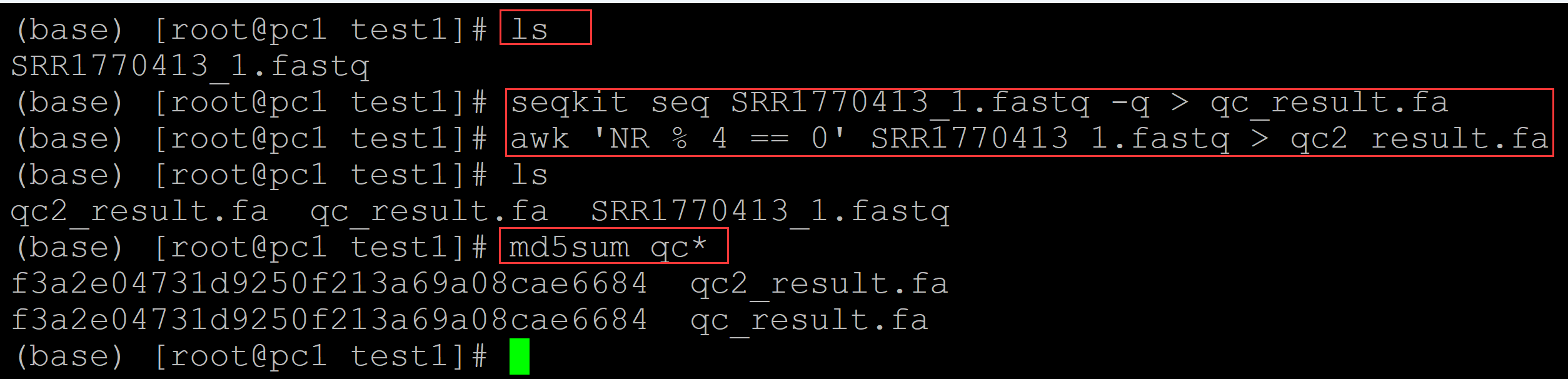
015、提取碱基的质量值
(base) [root@pc1 test1]# ls SRR1770413_1.fastq (base) [root@pc1 test1]# seqkit seq SRR1770413_1.fastq -q > qc_result.fa ## 提取碱基质量值 (base) [root@pc1 test1]# awk 'NR % 4 == 0' SRR1770413_1.fastq > qc2_result.fa ## 提取碱基质量值 (base) [root@pc1 test1]# ls qc2_result.fa qc_result.fa SRR1770413_1.fastq (base) [root@pc1 test1]# md5sum qc* ## 比较结果 f3a2e04731d9250f213a69a08cae6684 qc2_result.fa f3a2e04731d9250f213a69a08cae6684 qc_result.fa
016、去除序列gap
(base) [root@pc1 test1]# ls test.fna (base) [root@pc1 test1]# echo -e ">seq\nACGT-actgc-ACC" ## 原始序列 >seq ACGT-actgc-ACC (base) [root@pc1 test1]# echo -e ">seq\nACGT-actgc-ACC" | seqkit seq -g -u ## 去除序列gap >seq ACGTACTGCACC

。



【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· 震惊!C++程序真的从main开始吗?99%的程序员都答错了
· 【硬核科普】Trae如何「偷看」你的代码?零基础破解AI编程运行原理
· 单元测试从入门到精通
· 上周热点回顾(3.3-3.9)
· winform 绘制太阳,地球,月球 运作规律
2022-09-25 pindel软件的基本用法