
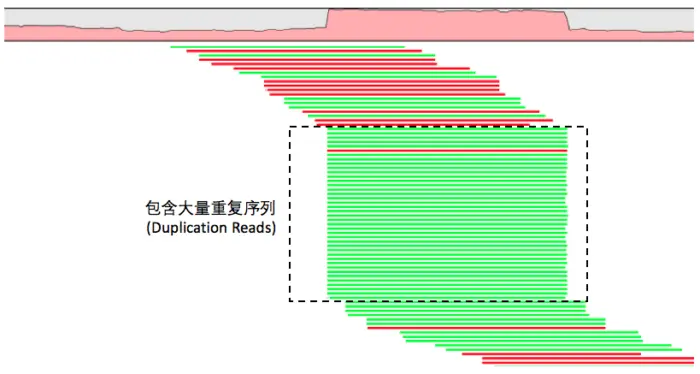
bam文件去重复
建库过程PCA扩增过程中引入重复序列,会对变异检测结果产生影响,重复的DNA片段会比对到参考基因组的相同位置,根据这一特点来进行去重复。
001、gatk(picard标记重复)
gatk MarkDuplicates -I sample01.sorted.bam -O sample01.sorted.markdup.bam -M sample01.sorted.markdup_metrics.txt
002、samtools
samtools sort -n xxx.bam -o xxx.sort.bam ## step1,按read name排序 samtools fixmate -m xxx.sort.bam xxx.fixmate.bam ## step2 samtools sort xxx.fixmate.bam -o xxx.positionsort.bam ## step3,按position排序 samtools markdup -r xxx.positionsort.bam xxx.markdup.bam ## step4,加上-r就会直接去掉重复序列
## 或者合并命令: samtools sort -n xxx.bam | samtools fixmate -m | samtools sort | samtools markdup -r > xxx.markdup.bam
参考:
01、https://www.jianshu.com/p/8cdbb89530c6?utm_campaign=maleskine&utm_content=note&utm_medium=seo_notes
02、https://www.cnblogs.com/bio-mary/p/12053346.html
03、https://www.jianshu.com/p/e20a3b73dcd0
。
.



【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· 震惊!C++程序真的从main开始吗?99%的程序员都答错了
· 【硬核科普】Trae如何「偷看」你的代码?零基础破解AI编程运行原理
· 单元测试从入门到精通
· 上周热点回顾(3.3-3.9)
· winform 绘制太阳,地球,月球 运作规律
2022-09-22 linux 中统计相同序列出现的次数
2022-09-22 sbatch命令在集群递交任务模板
2022-09-22 linux 中 利用命令向文件的末尾添加空行
2022-09-22 linux中basename命令
2022-09-22 linux 中 date +%s 获取1970年以来的秒数