二代测序分析流程
001、
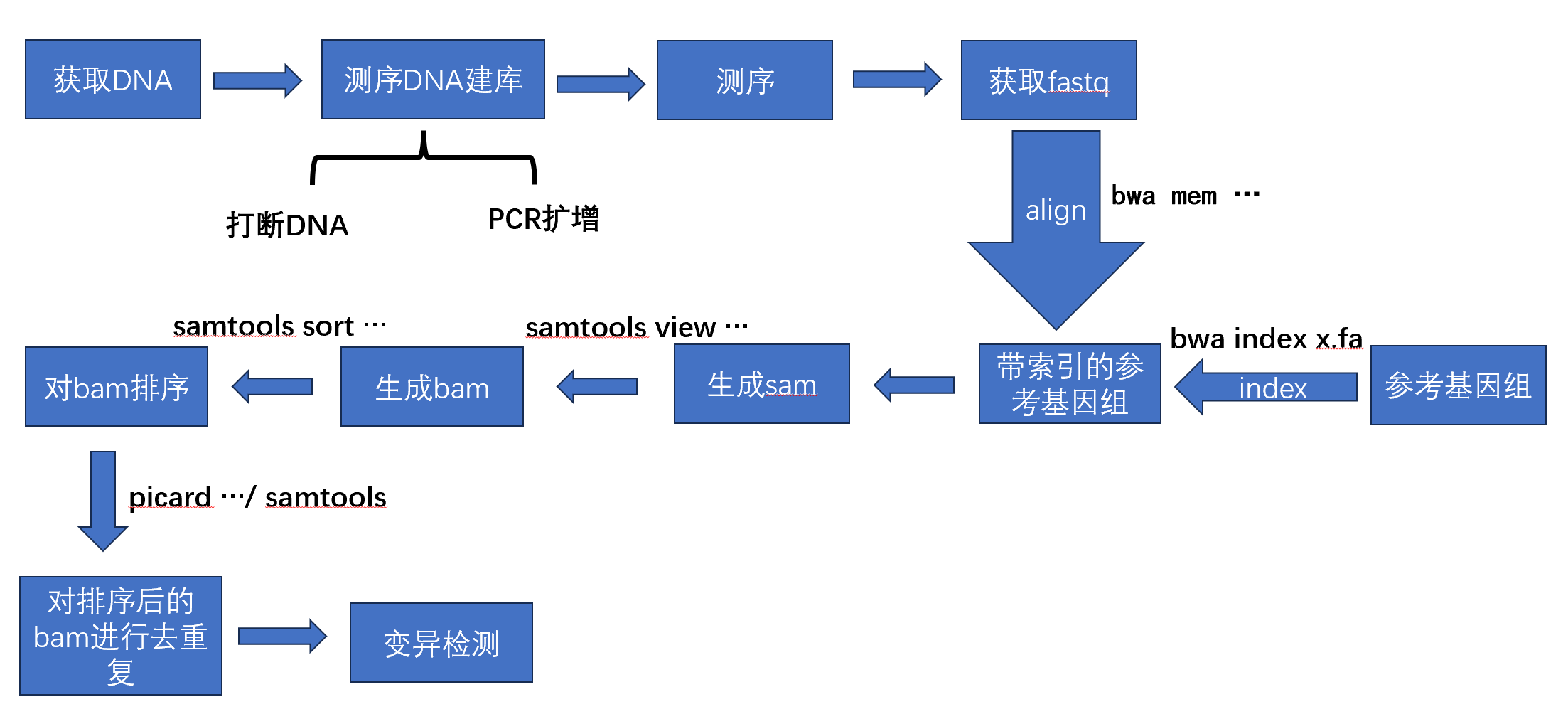
01、bwa index x.fa:
对参考基因组构建索引,这其实是在为参考序列进行Burrows Wheeler变换(wiki: 块排序压缩),以便能够在序列比对的时候进行快速的搜索和定位。
02、samtools sort:
因为测序的reads是无序的,比对生成的sam/bam文件也是无序的。
03、去重复
重复DNA片段的来源:建库过程中PCR的扩增
重复DNA片段的影响:对结果造成偏差,造成假阳性和假阴性。
1、DNA片段打断的过程中,会发生转换或者颠换,PCR反应会放大这种错误(假阳性)
2、PCR反应过程自身也会产生错误(假阳性)
3、PCR bias:某些DNA片段PCR反应强烈,如果含有reference序列的片段反应强烈,则会掩盖掉真实的变异(假阴性)
去重复的原理:PCR扩增的片段都是一样的,那么比对到参考基因组的位置也是一样的(原始的DNA片段,长度完全一样概率低),可以根据这个特点找到这些重复序列了。
04、
参考:
01、https://www.jianshu.com/p/8cdbb89530c6?utm_campaign=maleskine&utm_content=note&utm_medium=seo_notes

 浙公网安备 33010602011771号
浙公网安备 33010602011771号