
linux 中 shell脚本实现提取gff文件中的最长转录本
001、数据(山羊ARS1的gff文件)
脚本 如下:
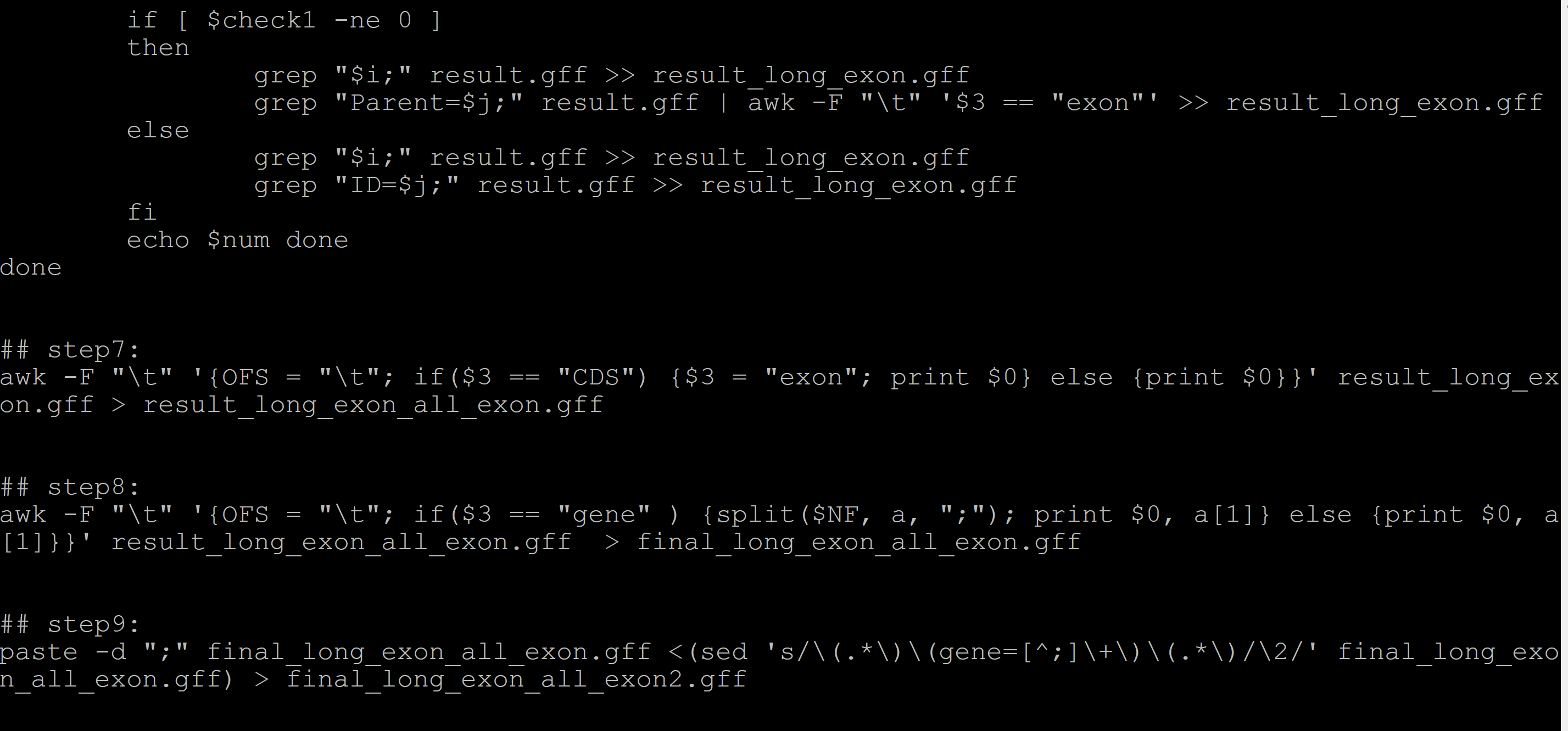
(base) [b20223040323@admin2 001_standard_gff_file]$ cat record.sh #!/bin/bin ## step1: eliminate the influence of pseudogene grep -v "^#" GCF_001704415.1_ARS1_genomic.gff | awk -F "\t" 'BEGIN{tag = "yes"}{if($3 == "pseudogene") {tag = "no"}; if($3 == "gene") {tag = "yes"}; if(tag == "yes") {print $0}}' > result.gff ## step2: exclude the genes without exon awk -F "\t" 'BEGIN{sum = 0}{if($3 == "gene"){sum++} else {sum = 0}; if(sum > 1) {print NR - 1}}' result.gff | while read i do sed -i "$i s/^/detele_tag\t/" result.gff done sed -i '/detele_tag\t/d' result.gff ## step3: num=0 awk -F "\t" '$3 == "gene"{print $NF}' result.gff | cut -d ";" -f 1 | while read i do ## 提取基因ID let num=$num+1 check1=$(grep "$i;" result.gff | wc -l) if [ $check1 -ne 1 ] then echo "$i layer1 check" exit fi grep "$i;" result.gff >> 001.gff ## 基因ID 写入文件 str1=$(grep "$i;" result.gff | awk -F "\t" '{print $NF}' | cut -d ";" -f 1 | cut -d "=" -f 2) check2=$(grep "Parent=$str1;" result.gff | wc -l) echo "$i: $check2" >> 002.txt ## 记录每个基因的转录本数目 if [ $check2 -lt 1 ] then echo "$i layer 2 check" exit fi grep "Parent=$str1;" result.gff > tmp for j in $(seq $check2) do ## 提取每一个转录本的ID sed -n "$j"p tmp >> 001.gff ## 转录本ID写入文件 str2=$(sed -n "$j"p tmp | awk -F "\t" '{print $NF}' | cut -d ";" -f 1 | cut -d "=" -f 2) check3=$(grep "Parent=$str2;" result.gff | wc -l) if [ $check3 -eq 0 ] then echo -e "$i\t$str2\thave no exon" >> gene_trans_no_exon.gff sed -n "$j"p tmp | awk -v a=$i -v b=$str2 'BEGIN{sum = 0} {OFS = "\t"; sum += ($5 - $4 + 1)} END {print a, b, sum}' >> 003.txt elif [ $check3 -gt 0 ] then check4=$(grep "Parent=$str2;" result.gff | awk -F "\t" '$3 == "exon"' | wc -l) if [ $check4 -gt 0 ] then grep "Parent=$str2;" result.gff | awk -F "\t" '$3 == "exon"' >> 001.gff ## 外显子写入文件 grep "Parent=$str2;" result.gff | awk -F "\t" '$3 == "exon"' | awk -v a=$i -v b=$str2 'BEGIN{sum = 0} {OFS = "\t"; sum += ($5 - $4 + 1)} END {print a, b, sum}' >> 003.txt else echo -e "$i\t$str2" >> check.txt grep "Parent=$str2;" result.gff | awk -v a=$i -v b=$str2 '{OFS = "\t"; print a, b, $0}' >> gene_trans_no_exon_have_other.gff grep "Parent=$str2;" result.gff >> 001.gff grep "Parent=$str2;" result.gff | awk -v a=$i -v b=$str2 'BEGIN{sum = 0} {OFS = "\t"; sum += ($5 - $4 + 1)} END {print a, b, sum}' >> 003.txt fi else continue fi done ## 统计每一个转录本的长度并写入文件 rm -f tmp echo $num done done ## step4: awk '{if(ay[$1] == "") {ay[$1] = $3}; if(ay[$1] < $3) {ay[$1] = $3}} END {OFS = "\t"; for (i in ay) {print i, ay[i]}}' 003.txt | while read {m,n} do awk -F "\t" -v a=$m -v b=$n '{if($1 == a && $3 == b) {print $0}}' 003.txt >> long_transcript_id_tmp.txt done ## step5: cut -f 1,3 long_transcript_id_tmp.txt | awk '{if(NR > 1 && $0 == tmp) {next} else {print NR}; tmp = $0}' | while read i do sed -n "$i"p long_transcript_id_tmp.txt >> long_transcript.txt done ## step6: num=0 cut -f 1-2 long_transcript.txt | while read {i,j} do let num=$num+1 check1=$(grep "Parent=$j;" result.gff | wc -l) if [ $check1 -ne 0 ] then grep "$i;" result.gff >> result_long_exon.gff grep "Parent=$j;" result.gff | awk -F "\t" '$3 == "exon"' >> result_long_exon.gff else grep "$i;" result.gff >> result_long_exon.gff grep "ID=$j;" result.gff >> result_long_exon.gff fi echo $num done done ## step7: awk -F "\t" '{OFS = "\t"; if($3 == "CDS") {$3 = "exon"; print $0} else {print $0}}' result_long_exon.gff > result_long_exon_all_exon.gff ## step8: awk -F "\t" '{OFS = "\t"; if($3 == "gene" ) {split($NF, a, ";"); print $0, a[1]} else {print $0, a[1]}}' result_long_exon_all_exon.gff > final_long_exon_all_exon.gff ## step9: paste -d ";" final_long_exon_all_exon.gff <(sed 's/\(.*\)\(gene=[^;]\+\)\(.*\)/\2/' final_long_exon_all_exon.gff) > final_long_exon_all_exon2.gff
。
 浙公网安备 33010602011771号
浙公网安备 33010602011771号