
samtools 统计重测序数据深度 depth、depth -a、depth -aa的联系与区别
测试数据链接:
链接:https://pan.baidu.com/s/18_R3W1K7DYdUA9DxGZ0jxw
提取码:nklh
--来自百度网盘超级会员V6的分享
背景知识:
将质控后的fastq文件比对到参考基因组之后,有可能会有部分染色体根本比对不上。 比如参考基因组中一共有10条染色体,结果fastq的reads仅比对到8条染色体,那么就是说有两条染色体压根就没有比对上。
用一个实际的测试数据来看一下:
这里准备了两个文件:一个参考基因组fasta文件,和一个按照一般流程将fastq的reads数据比对到参考基因组后生成的排序后的bam文件,利用这两个文件可以查看参考基因组一共有多少条染色体, fastq数据一共比对上多少条染色体。
统计参考基因组的染色体数目:
(base) [b20223040323@admin1 test]$ ls GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam (base) [b20223040323@admin1 test]$ grep "^>" GCF_000005845.2_ASM584v2_genomic.fna ## 统计参考基因组的染色体数目 >chr1 >chr2 >chr3
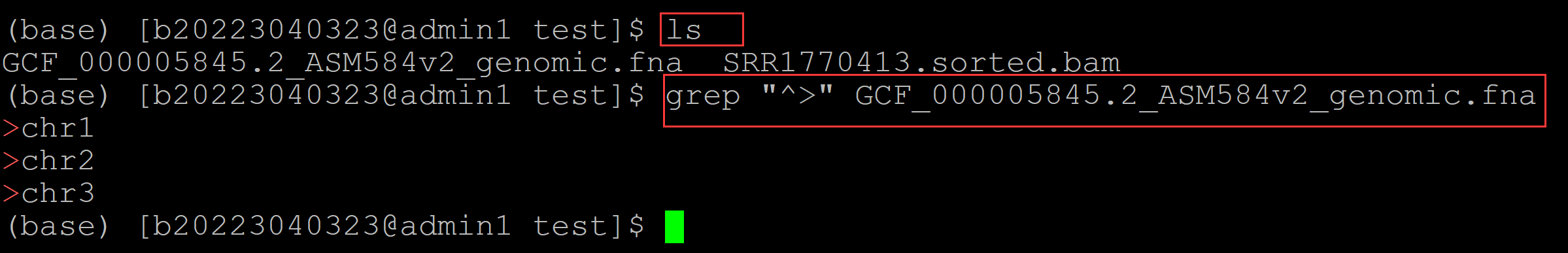
可见该参考基因组一共有三条染色体。
利用bam文件统计一共比对上多少条染色体:
(base) [b20223040323@admin1 test]$ ls GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam (base) [b20223040323@admin1 test]$ samtools view SRR1770413.sorted.bam | cut -f 3 | uniq ## 利用bam文件统计一共比对上多少条染色体 chr1 chr2 *
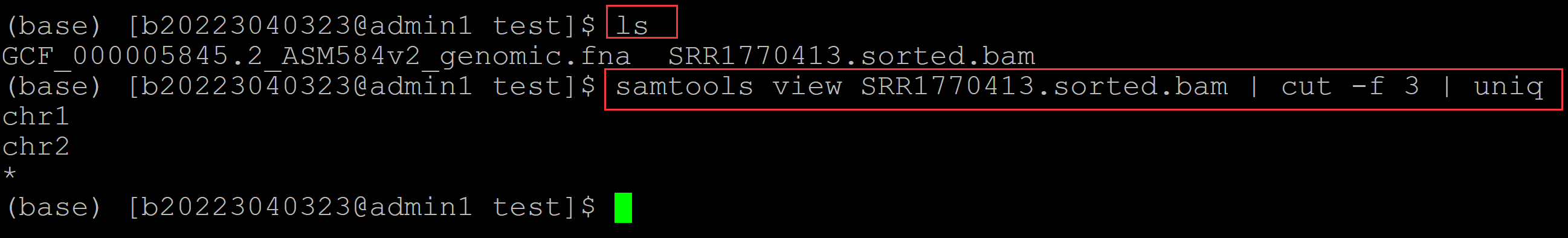
可见一共比对上的染色体数有两条,也就是说chr3并没有比对上。
比较samtools计算测序深度时:depth、depth -a、depth -aa的区别。
001、首先先统计一下参考基因组中每一条染色体的长度,可以使用samtools软件实现:

(base) [b20223040323@admin1 test]$ ls GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam (base) [b20223040323@admin1 test]$ samtools faidx GCF_000005845.2_ASM584v2_genomic.fna ### 统计每一条染色体的长度 (base) [b20223040323@admin1 test]$ ls GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam GCF_000005845.2_ASM584v2_genomic.fna.fai (base) [b20223040323@admin1 test]$ cat GCF_000005845.2_ASM584v2_genomic.fna.fai ## 查看,第二列是每一条染色体的长度 chr1 4641652 6 80 81 chr2 39862 4699685 80 81 chr3 398 4740052 80 81
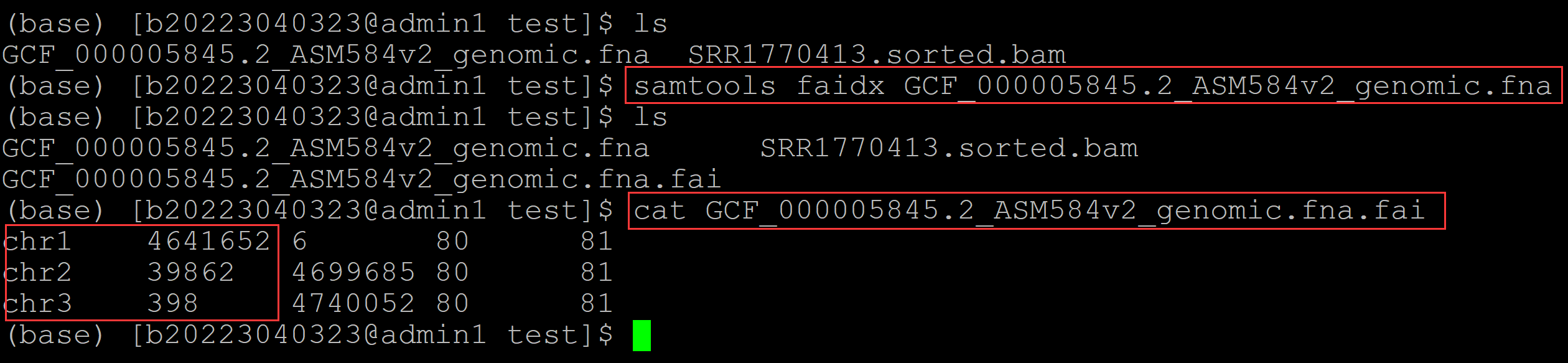
可见chr1的长度为:4641652。。。。。
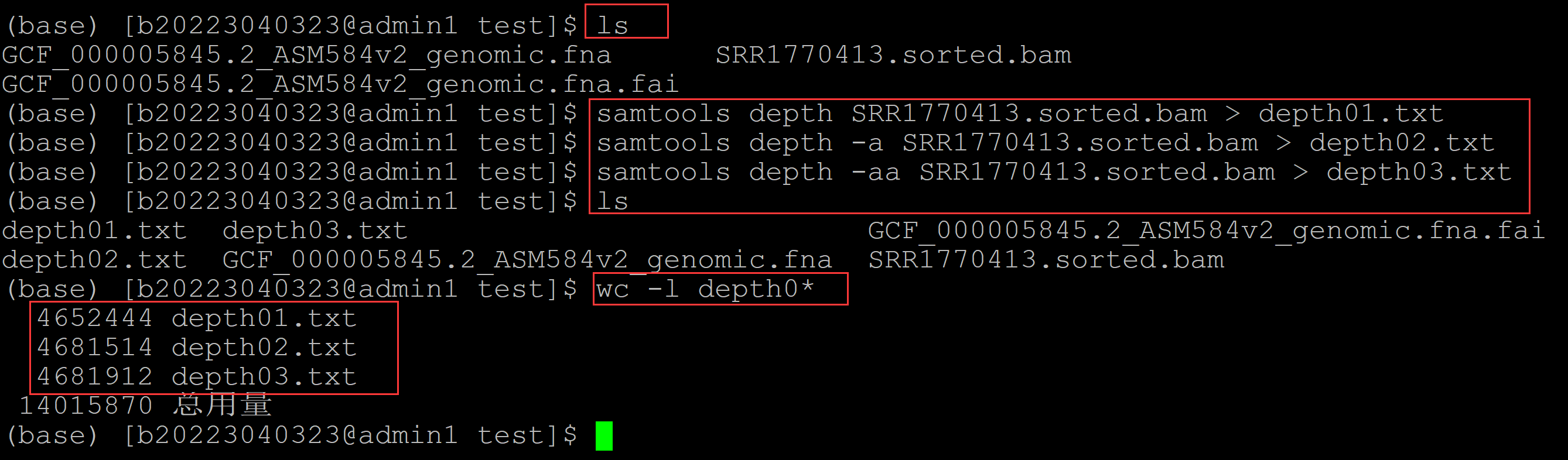
002、利用samtools的depth、depth -a、depth -aa分别对同一个bam文件进行测序深度统计
(base) [b20223040323@admin1 test]$ ls GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam GCF_000005845.2_ASM584v2_genomic.fna.fai (base) [b20223040323@admin1 test]$ samtools depth SRR1770413.sorted.bam > depth01.txt ## depth (base) [b20223040323@admin1 test]$ samtools depth -a SRR1770413.sorted.bam > depth02.txt ## depth -a (base) [b20223040323@admin1 test]$ samtools depth -aa SRR1770413.sorted.bam > depth03.txt ## depth -aa (base) [b20223040323@admin1 test]$ ls depth01.txt depth03.txt GCF_000005845.2_ASM584v2_genomic.fna.fai depth02.txt GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam (base) [b20223040323@admin1 test]$ wc -l depth0* ## 统计生成的depth文件的行数,可见三个文件行数不一致 4652444 depth01.txt 4681514 depth02.txt 4681912 depth03.txt 14015870 总用量
003、samtools的depth参数统计的是比对到参考基因组的染色体上所有位点测序深度大于0的所有测序深度数据,用一下脚本简单验证:
(base) [b20223040323@admin1 test]$ ls depth01.txt depth03.txt GCF_000005845.2_ASM584v2_genomic.fna.fai depth02.txt GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam (base) [b20223040323@admin1 test]$ head -n 5 depth01.txt ## depth01.txt文件记录的是位点的测序深度,每行一个位点 chr1 1 12 chr1 2 13 chr1 3 14 chr1 4 15 chr1 5 15 (base) [b20223040323@admin1 test]$ awk '{print $3}' depth01.txt | sort -n | head -n 3 ## 对第三列深度信息进行排序,可见最小深度为1 1 1 1 (base) [b20223040323@admin1 test]$ awk '{print $3}' depth01.txt | sort -n | tail -n 3 ## 输出最大测序深度 509 510 510
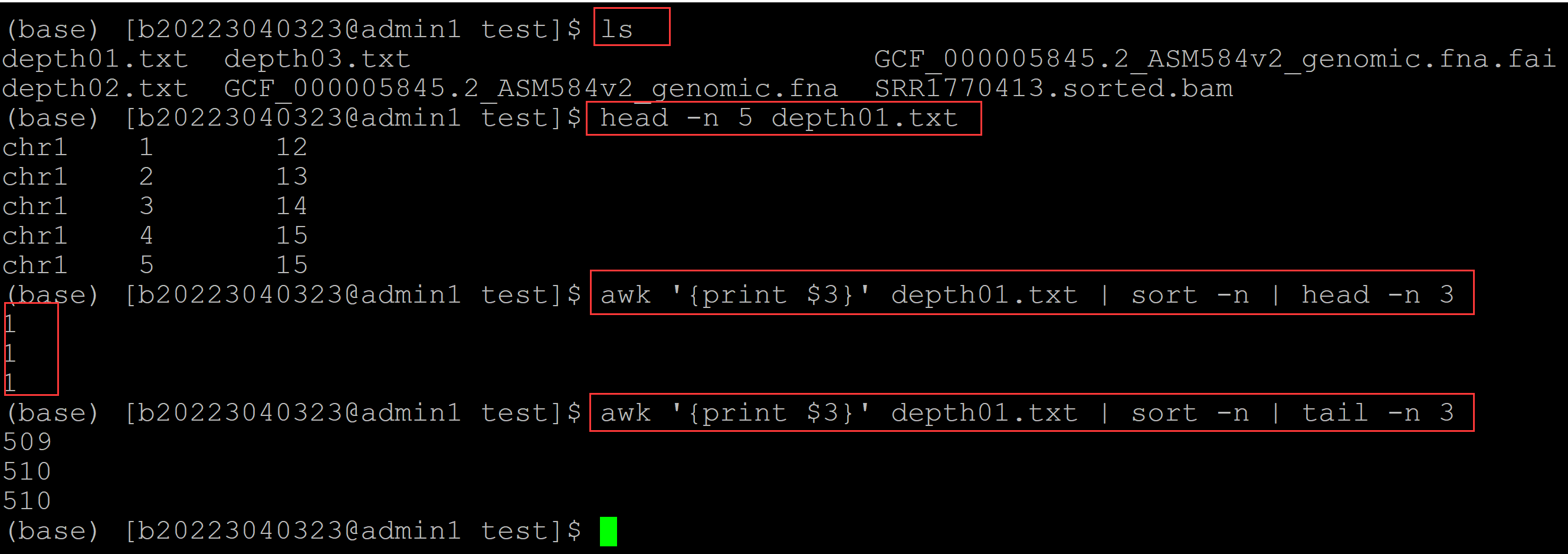
可见 samtools depth参数输出测序深度的结果最小的测序深度为1.
004、samtools depth -a参数输出的结果为fastq比对到参考基因组的染色体上的所有位点的测序深度,即在depth参数输出结果的基础上,也输出测序深度为0的位点。可用如下脚本进行验证:
a、如果输出fastq数据比对到参考基因组的染色体上所有位点的测序深度, 那么depth02.txt的行数应该等于比对到染色体长度的总和,即chr1和chr2的总长度。
(base) [b20223040323@admin1 test]$ ls depth01.txt depth03.txt GCF_000005845.2_ASM584v2_genomic.fna.fai depth02.txt GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam (base) [b20223040323@admin1 test]$ wc -l depth02.txt 4681514 depth02.txt (base) [b20223040323@admin1 test]$ cat GCF_000005845.2_ASM584v2_genomic.fna.fai chr1 4641652 6 80 81 chr2 39862 4699685 80 81 chr3 398 4740052 80 81 (base) [b20223040323@admin1 test]$ awk 'NR <= 2 {sum += $2} END { print sum }' GCF_000005845.2_ASM584v2_genomic.fna.fai ##统计比对到的染色体的总长度 4681514
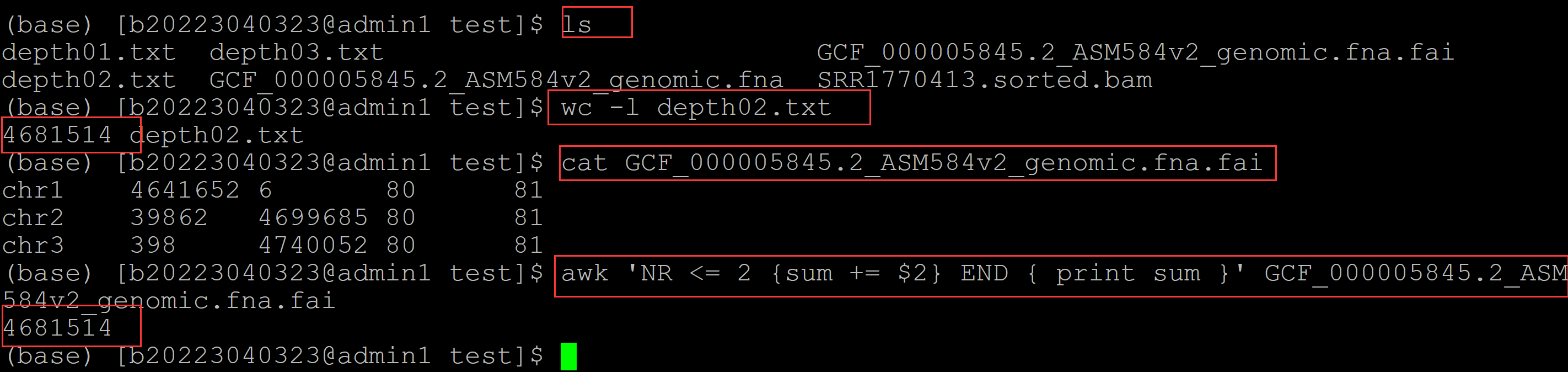
可以看到depth02.txt的行数等于比对到的染色体的总长度。
b、如果depth -a参数是在depth的基础上增加输出了测序深度为0的位点,那么depth02.txt 和 depth01.txt相比,相同的部分和depth01.txt完全一致, 不同的位点的测序深度都为0.
(base) [b20223040323@admin1 test]$ ls depth01.txt depth03.txt GCF_000005845.2_ASM584v2_genomic.fna.fai depth02.txt GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam (base) [b20223040323@admin1 test]$ sort depth01.txt | md5sum ## depth01.txt排序,并计算MD5 53c631aeb9024f330280bb3762310189 - (base) [b20223040323@admin1 test]$ cat depth01.txt depth02.txt | sort | uniq -d | md5sum ## 去depth01.txt和depth02.txt的交集并计算MD5 53c631aeb9024f330280bb3762310189 - (base) [b20223040323@admin1 test]$ cat depth01.txt depth01.txt depth02.txt | sort | uniq -u | awk '{print $3}' | uniq -c ## 查看depth02.txt中多处的行,并查看测序深度 29070 0
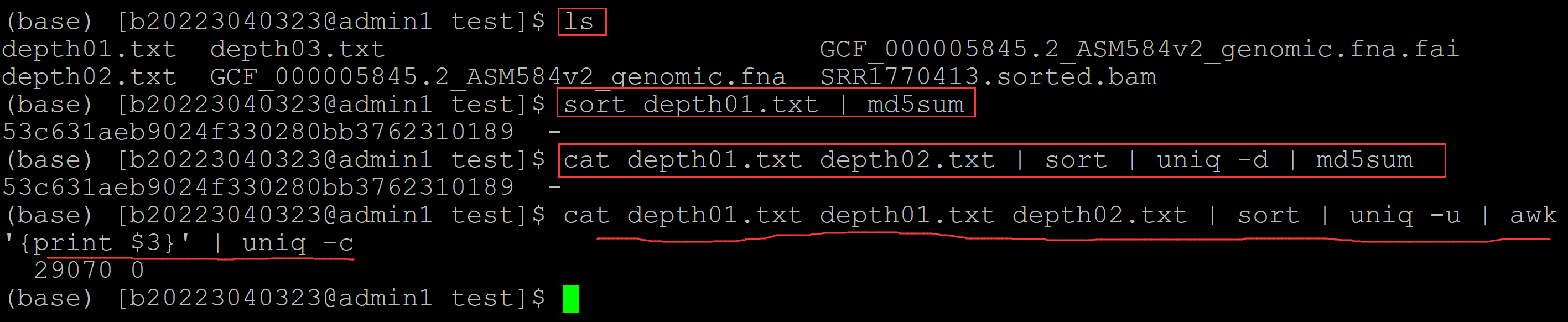
可以看到depth01.txt和depth02.txt中的重合部分和depth01.txt完全一致,同时depth02.txt中多出的部分的测序深度都为0.
005、samtools depth -aa参数输出fasta文件中所有染色体上的位点的测序深度,包括没有比对上的染色体的位点上的测序深度。
a、如果depth -aa输出fasta文件中所有染色体上的位点的测序深度,那么depth03.txt的行数等于fasta文件中所有染色体的总长度:
(base) [b20223040323@admin1 test]$ ls depth01.txt depth03.txt GCF_000005845.2_ASM584v2_genomic.fna.fai depth02.txt GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam (base) [b20223040323@admin1 test]$ wc -l depth03.txt ## 统计行数 4681912 depth03.txt (base) [b20223040323@admin1 test]$ cat GCF_000005845.2_ASM584v2_genomic.fna.fai chr1 4641652 6 80 81 chr2 39862 4699685 80 81 chr3 398 4740052 80 81 (base) [b20223040323@admin1 test]$ awk '{sum += $2} END {print sum}' GCF_000005845.2_ASM584v2_genomic.fna.fai ## 统计所有染色体的总长度 4681912
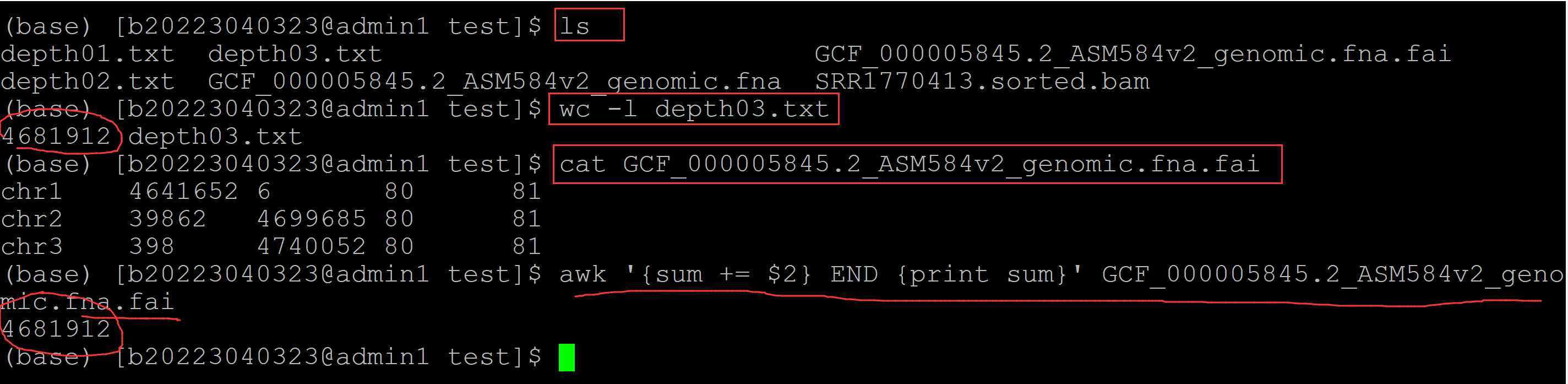
可以看到depth03.txt的长度等于fasta文件中所有染色体的总长度。
b、depth03.txt和depth02.txt相比,如果多出的是没有比对上的染色体的总长度,那么depth03.txt比depth02.txt多出没有比对到的染色体的长度,且多出位点的测序深度均为0.
(base) [b20223040323@admin1 test]$ ls depth01.txt depth03.txt GCF_000005845.2_ASM584v2_genomic.fna.fai depth02.txt GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam (base) [b20223040323@admin1 test]$ echo "$(wc -l < depth03.txt) - $(wc -l < depth02.txt)" | bc ## 计算行数只差 398 (base) [b20223040323@admin1 test]$ cat GCF_000005845.2_ASM584v2_genomic.fna.fai chr1 4641652 6 80 81 chr2 39862 4699685 80 81 chr3 398 4740052 80 81 (base) [b20223040323@admin1 test]$ cat depth02.txt depth02.txt depth03.txt | sort | uniq -u | awk '{print $3}' | uniq -c ## 输出depth03.txt相对于depth02.txt多出的位点的测序深度信息 398 0
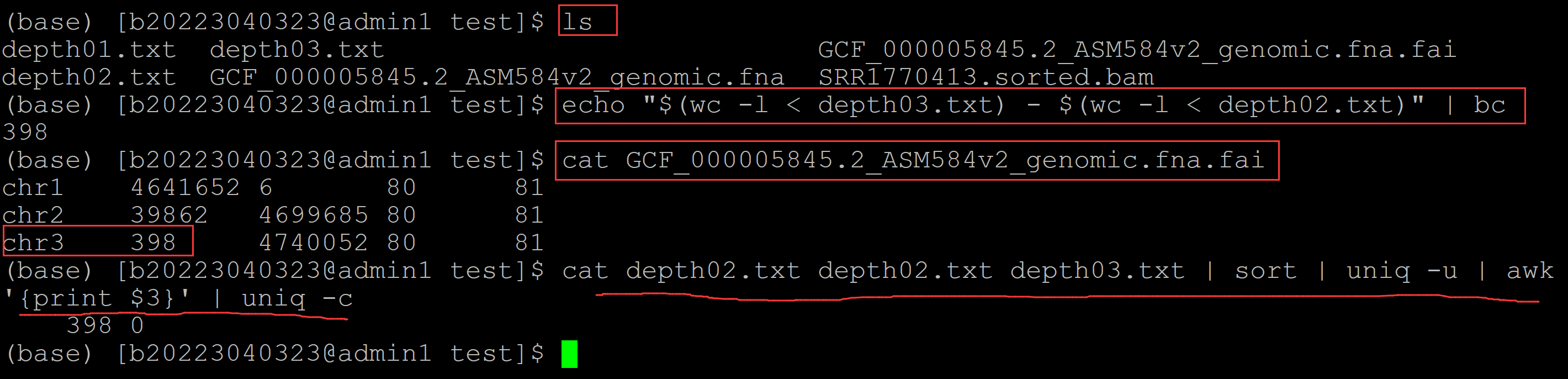
c、depth03.txt 和 depth01.txt相对,多出额外的一条染色体的测序深度信息,且测序深度均为0.
(base) [b20223040323@admin1 test]$ ls depth01.txt depth03.txt GCF_000005845.2_ASM584v2_genomic.fna.fai depth02.txt GCF_000005845.2_ASM584v2_genomic.fna SRR1770413.sorted.bam (base) [b20223040323@admin1 test]$ cut -f 1 depth01.txt | uniq ## 输出染色体 chr1 chr2 (base) [b20223040323@admin1 test]$ cut -f 1 depth03.txt | uniq ## 输出染色体 chr1 chr2 chr3 (base) [b20223040323@admin1 test]$ cat depth01.txt depth01.txt depth03.txt | sort | uniq -u | awk '{print $3}' | uniq -c ## 输出depth03.txt相对于depth01.txt额外的位点的深度信息 29468 0
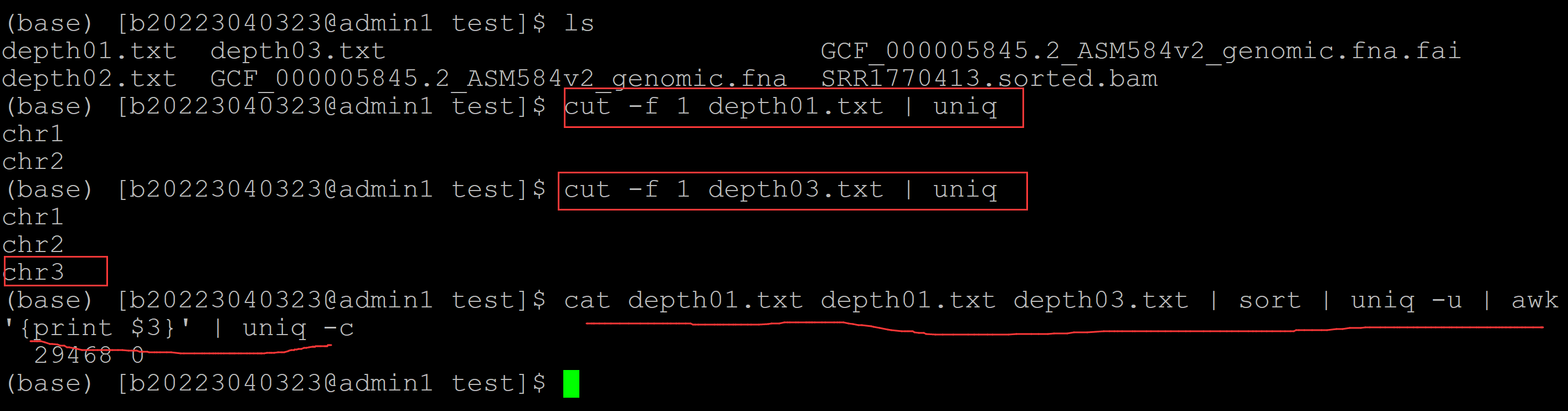
可以看到depth03.txt先对于depth01.txt多出了一条染色体,且多出的位点的测序深度均为0.
小结:
samptools depth: 输出fastq数据比对到染色体的位点深度大于0的深度信息。
samtools depth -a:输出fastq数据比对到染色体的所有位点的测序深度信息,包含0深度位点。
samtools depth -aa:输出fasta文件中所有染色体上位点的深度信息,包含fastq数据没有比对到的染色体。



【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· 震惊!C++程序真的从main开始吗?99%的程序员都答错了
· 【硬核科普】Trae如何「偷看」你的代码?零基础破解AI编程运行原理
· 单元测试从入门到精通
· 上周热点回顾(3.3-3.9)
· winform 绘制太阳,地球,月球 运作规律
2021-03-08 python中如何获取函数文档
2021-03-08 python中写一个求阶乘的函数
2021-03-08 python中写一个求阶乘的函数
2021-03-08 python中map()内置函数
2021-03-08 python中filter关键字
2021-03-08 python中lambda关键字创建匿名函数
2021-03-08 python中lambda关键字--定义匿名函数