
python 中统计fasta文件中每条scaffold中碱基的数目
001、

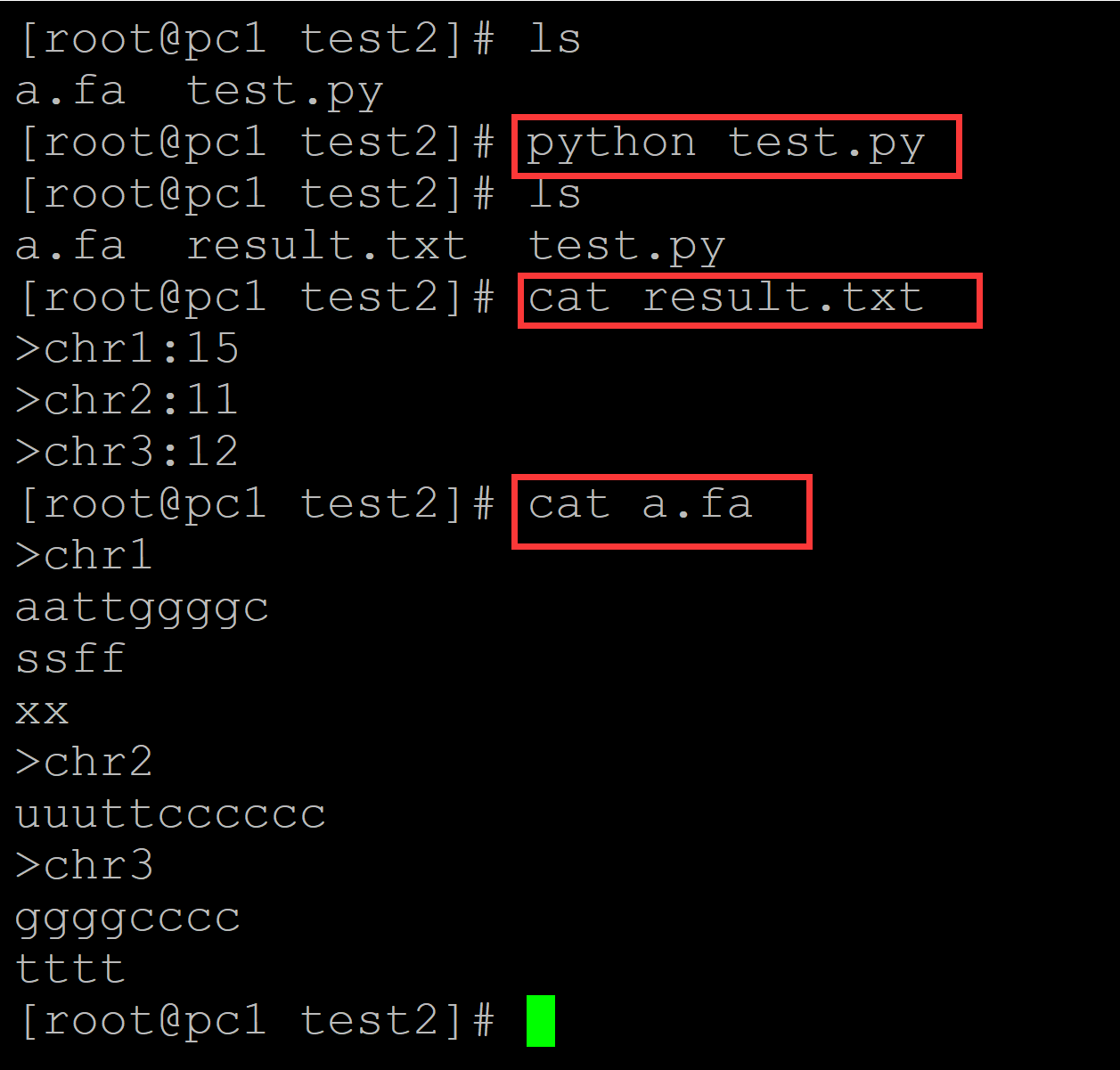
[root@pc1 test2]# ls a.fa test.py [root@pc1 test2]# cat a.fa ## 测试fasta文件 >chr1 aattggggc ssff xx >chr2 uuuttcccccc >chr3 ggggcccc tttt [root@pc1 test2]# cat test.py ## 统计脚本 #!/usr/bin/python in_file = open("a.fa", "r") out_file = open("result.txt", "w") dict1 = dict() for i in in_file: i = i.strip() if i.startswith(">"): key = i dict1[key] = str() else: dict1[key] += i for i,j in dict1.items(): out_file.write(i + ":") print(len(j), file = out_file) in_file.close() out_file.close()
[root@pc1 test2]# ls a.fa test.py [root@pc1 test2]# python test.py ## 执行程序 [root@pc1 test2]# ls a.fa result.txt test.py [root@pc1 test2]# cat result.txt ## 执行结果 >chr1:15 >chr2:11 >chr3:12 [root@pc1 test2]# cat a.fa ## 原始文件 >chr1 aattggggc ssff xx >chr2 uuuttcccccc >chr3 ggggcccc tttt
002、改进速度
[root@pc1 test1]# cat test.py #!/usr/bin/python in_file = open("a.fa", "r") out_file = open("result.txt", "w") dict1 = dict() for i in in_file: i = i.strip() if i.startswith(">"): key = i dict1[key] = list() else: dict1[key].append(i) for i,j in dict1.items(): out_file.write(i.split(" ")[0] + ":") length = 0 for k in j: length += len(k) print(length, file = out_file) in_file.close() out_file.close()
[root@pc1 test1]# ls a.fa test.py [root@pc1 test1]# ll -h ## 测试1G参考基因组 total 1018M -rw-r--r--. 1 root root 1018M Nov 12 20:46 a.fa -rw-r--r--. 1 root root 391 Nov 12 20:51 test.py [root@pc1 test1]# time python test.py ## 测试程序 real 0m11.972s user 0m9.793s sys 0m2.179s [root@pc1 test1]# ls a.fa result.txt test.py



【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· 震惊!C++程序真的从main开始吗?99%的程序员都答错了
· 【硬核科普】Trae如何「偷看」你的代码?零基础破解AI编程运行原理
· 单元测试从入门到精通
· 上周热点回顾(3.3-3.9)
· winform 绘制太阳,地球,月球 运作规律
2020-11-12 ansible