
R语言中使用prcomp函数对单细胞数据进行PCA分析
前期参考:https://www.jianshu.com/p/4f7aeae81ef1
001、
library(dplyr) library(Seurat) library(patchwork) pbmc.data <- Read10X(data.dir = "C:/Users/75377/Desktop/r_test2/hg19") pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200) pbmc pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-") VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3) plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt") plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA") plot1 + plot2 pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5) pbmc <- NormalizeData(pbmc) pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000) top10 <- head(VariableFeatures(pbmc), 10) top10 plot1 <- VariableFeaturePlot(pbmc) plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE) all.genes <- rownames(pbmc) pbmc <- ScaleData(pbmc, features = all.genes)
002、利用R函数prcomp对单细胞数据进行PCA分析
genes = VariableFeatures(object = pbmc) length(genes) head(genes) dat <- pbmc[["RNA"]]@scale.data[genes,] ## pca分析用到的数据 dat <- t(dat) pca <- prcomp(dat,center = F,scale. = F) ## pca分析 plot(pca$x[,1], pca$x[,2], pch = 19) ## 利用前两个主成分绘图
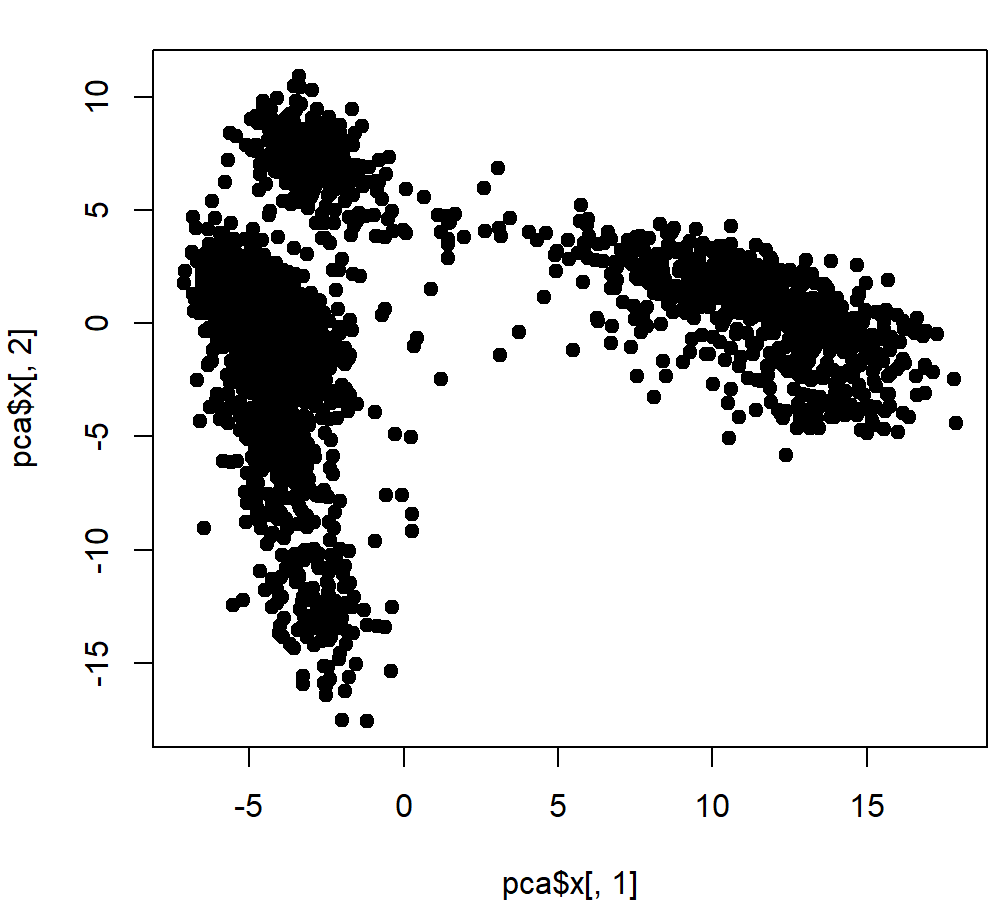
003、标准答案


 浙公网安备 33010602011771号
浙公网安备 33010602011771号