
Seurat4.0单细胞数据分析 数据的归一化
001、数据的归一化是在数据的标准化的基础上进行的,而且是按照行来进行的, 即:
(每一行的观测值 - 每一行的平均值)/每一行的标准差
验证:
a、前期步骤参考:https://www.jianshu.com/p/4f7aeae81ef1
b、

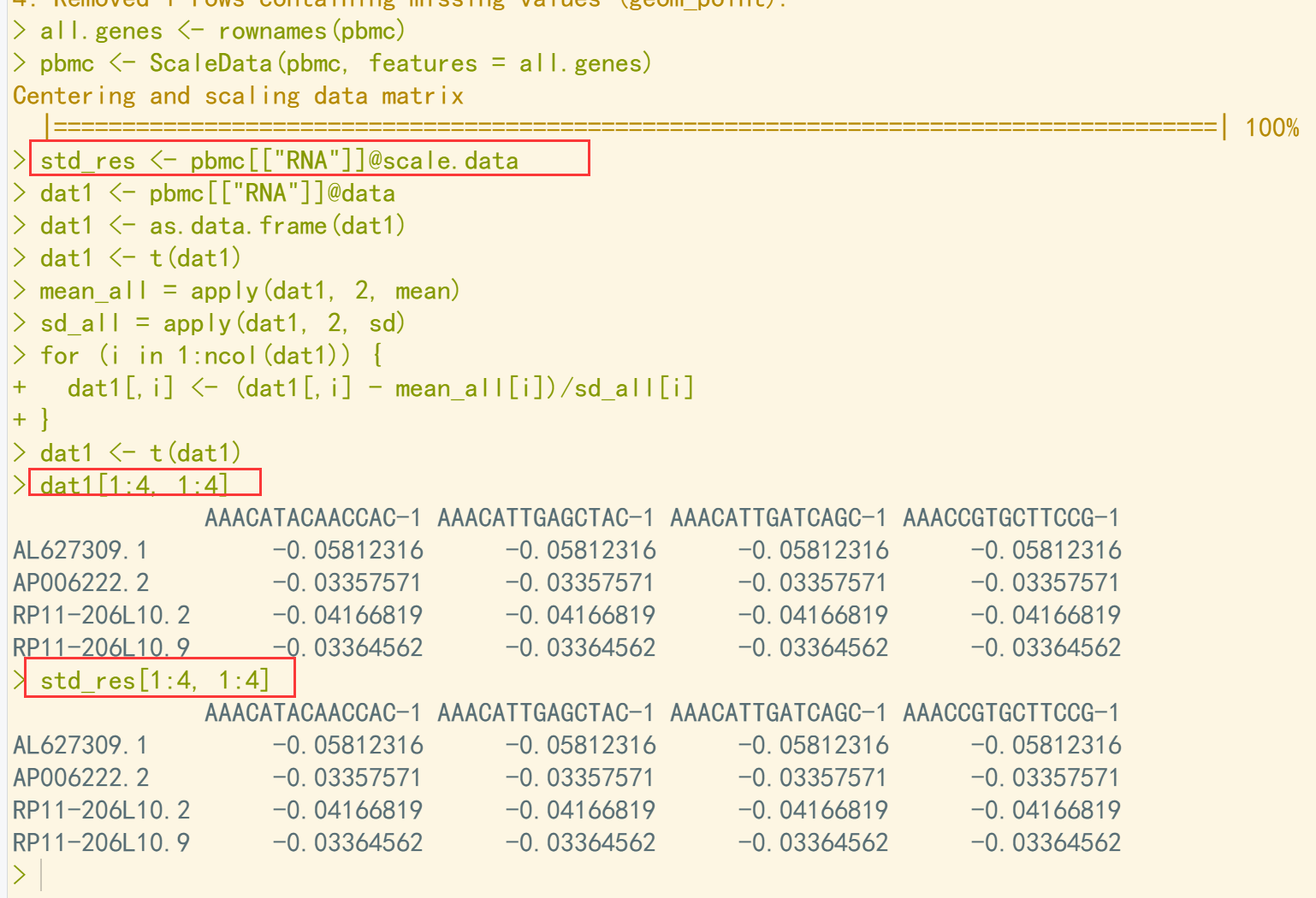
all.genes <- rownames(pbmc) pbmc <- ScaleData(pbmc, features = all.genes) std_res <- pbmc[["RNA"]]@scale.data ## 归一化标准答案 dat1 <- pbmc[["RNA"]]@data ## 在标准化数据的基础上进行归一化处理 dat1 <- as.data.frame(dat1) dat1 <- t(dat1) ## 首先转置, 按列计算速度快 mean_all = apply(dat1, 2, mean) sd_all = apply(dat1, 2, sd) for (i in 1:ncol(dat1)) { dat1[,i] <- (dat1[,i] - mean_all[i])/sd_all[i] ## 观测值减去平均值, 然后除以标准差 } dat1 <- t(dat1) ## 归一化计算结果 dat1[1:4, 1:4] std_res[1:4, 1:4] ## 对比结果



【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· 震惊!C++程序真的从main开始吗?99%的程序员都答错了
· 【硬核科普】Trae如何「偷看」你的代码?零基础破解AI编程运行原理
· 单元测试从入门到精通
· 上周热点回顾(3.3-3.9)
· winform 绘制太阳,地球,月球 运作规律