
R 脚本实现 xx.hmp.txt格式数据转换为plink格式
001、

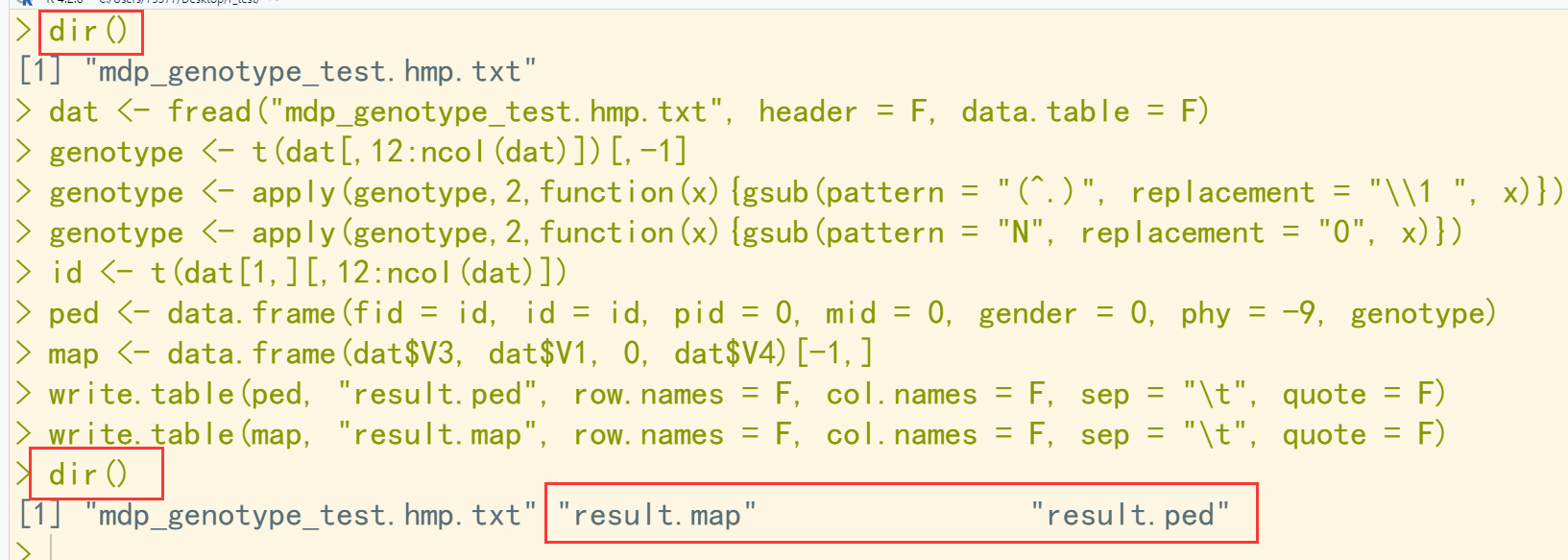
dir() dat <- fread("mdp_genotype_test.hmp.txt", header = F, data.table = F) ## 读入文件 genotype <- t(dat[,12:ncol(dat)])[,-1] genotype <- apply(genotype,2,function(x){gsub(pattern = "(^.)", replacement = "\\1 ", x)}) genotype <- apply(genotype,2,function(x){gsub(pattern = "N", replacement = "0", x)}) id <- t(dat[1,][,12:ncol(dat)]) ped <- data.frame(fid = id, id = id, pid = 0, mid = 0, gender = 0, phy = -9, genotype) map <- data.frame(dat$V3, dat$V1, 0, dat$V4)[-1,] write.table(ped, "result.ped", row.names = F, col.names = F, sep = "\t", quote = F) write.table(map, "result.map", row.names = F, col.names = F, sep = "\t", quote = F) dir()
002、plink验证
root@PC1:/home/test# ls result.map result.ped root@PC1:/home/test# plink --file result --recode PLINK v1.90b6.24 64-bit (6 Jun 2021) www.cog-genomics.org/plink/1.9/ (C) 2005-2021 Shaun Purcell, Christopher Chang GNU General Public License v3 Logging to plink.log. Options in effect: --file result --recode 15969 MB RAM detected; reserving 7984 MB for main workspace. .ped scan complete (for binary autoconversion). Warning: Variant 424 quadallelic; setting rarest alleles missing. Performing single-pass .bed write (3093 variants, 281 people). --file: plink-temporary.bed + plink-temporary.bim + plink-temporary.fam written. 3093 variants loaded from .bim file. 281 people (0 males, 0 females, 281 ambiguous) loaded from .fam. Ambiguous sex IDs written to plink.nosex . Using 1 thread (no multithreaded calculations invoked). Before main variant filters, 281 founders and 0 nonfounders present. Calculating allele frequencies... done. Total genotyping rate is 0.963828. 3093 variants and 281 people pass filters and QC. Note: No phenotypes present. --recode ped to plink.ped + plink.map ... done.



【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· 震惊!C++程序真的从main开始吗?99%的程序员都答错了
· 【硬核科普】Trae如何「偷看」你的代码?零基础破解AI编程运行原理
· 单元测试从入门到精通
· 上周热点回顾(3.3-3.9)
· winform 绘制太阳,地球,月球 运作规律
2021-07-29 awk命令末尾数字的作用
2021-07-29 c语言中如何创建、存储、输出字符串、输出字符串的大小、字符串的长度