
R语言实现统计plink格式数据基因频率
1、

dir() dat <- read.table("outcome.ped") dat <- dat[,-(1:6)] loci <- data.frame() loci[1:(nrow(dat) * 2), 1] <- 1 for (i in 1:(ncol(dat)/2)) { loci <- cbind(loci, c(dat[, 2*i -1], dat[,2 * i])) } loci <- loci[, -1] colnames(loci) <- paste0("c",1:8) result <- data.frame() temp <- vector() for (i in 1:ncol(loci)) { locidat <- as.data.frame(table(loci[,i])) if (nrow(locidat) == 1 & locidat[1, 1] == 0 ) { temp <- c(0, 0, 0, 1) }else if (nrow(locidat) == 1 & locidat[1, 1] != 0 ){ temp <- c(0, 0, as.character(locidat[1, 1]), 1) }else if(nrow(locidat) == 2) { locidat <- locidat[order(locidat$Var1),] if (locidat[1, 1] == 0) { temp <- c(0,0, as.character(locidat[2,1]), 1) }else { locidat <- locidat[order(locidat$Freq),] temp <- c(as.character(locidat[1,1]), locidat[1,2]/sum(locidat$Freq), as.character(locidat[2,1]), locidat[2,2]/sum(locidat$Freq)) } }else if (nrow(locidat) == 3){ locidat <- locidat[locidat$Var1 != 0, ] locidat <- locidat[order(locidat$Freq),] temp <- c(as.character(locidat[1,1]),locidat[1,2]/sum(locidat$Freq), as.character(locidat[2,1]), locidat[2,2]/sum(locidat$Freq) ) } result <- rbind(result, temp) } for (i in c(2,4)) { result[,i] = round(as.numeric(result[,i]),5) } map <- read.table("outcome.map") result <- cbind(map[,c(1:2,4)], result) colnames(result) <- c("chr", "snp", "pos", "minor", "freq", "major", "freq") result write.csv(result, "result.csv", row.names = F) dir()

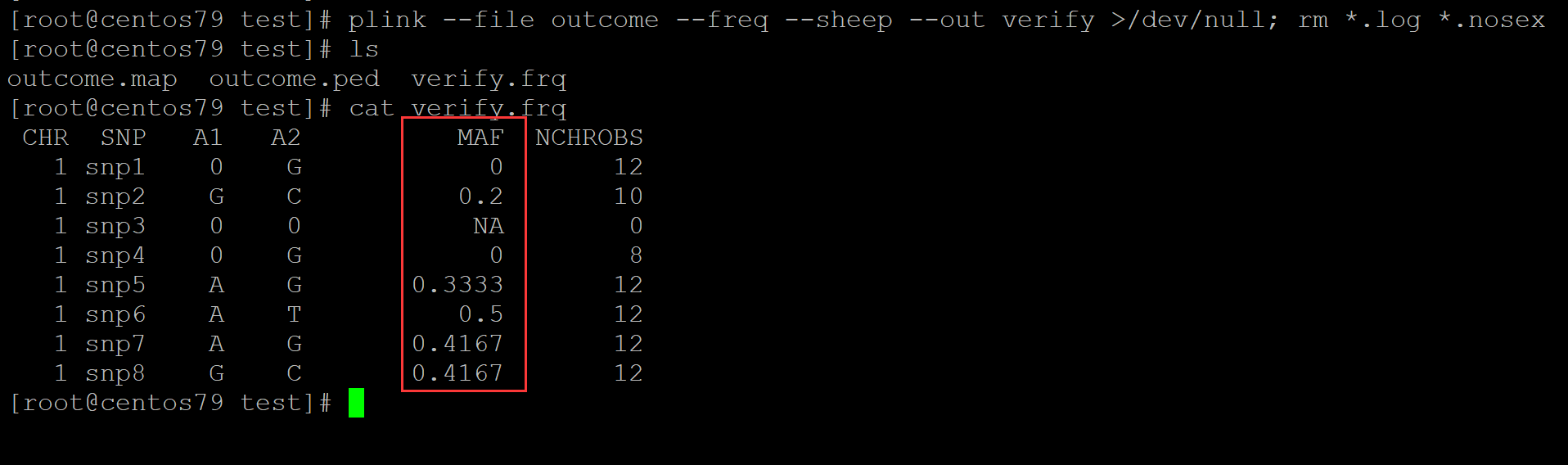
2、plink软件验证
[root@centos79 test]# plink --file outcome --freq --sheep --out verify >/dev/null; rm *.log *.nosex [root@centos79 test]# ls outcome.map outcome.ped verify.frq [root@centos79 test]# cat verify.frq CHR SNP A1 A2 MAF NCHROBS 1 snp1 0 G 0 12 1 snp2 G C 0.2 10 1 snp3 0 0 NA 0 1 snp4 0 G 0 8 1 snp5 A G 0.3333 12 1 snp6 A T 0.5 12 1 snp7 A G 0.4167 12 1 snp8 G C 0.4167 12



【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· 基于Microsoft.Extensions.AI核心库实现RAG应用
· Linux系列:如何用heaptrack跟踪.NET程序的非托管内存泄露
· 开发者必知的日志记录最佳实践
· SQL Server 2025 AI相关能力初探
· Linux系列:如何用 C#调用 C方法造成内存泄露
· 震惊!C++程序真的从main开始吗?99%的程序员都答错了
· 【硬核科普】Trae如何「偷看」你的代码?零基础破解AI编程运行原理
· 单元测试从入门到精通
· 上周热点回顾(3.3-3.9)
· winform 绘制太阳,地球,月球 运作规律
2020-10-30 linux系统中firewalld防火墙管理工具firewall-cmd(CLI命令行)
2020-10-30 linux系统中iptables防火墙管理工具