
使用detectRUNS包进行ROH检测,计算近交系数实践
1、测试数据下载
链接:https://pan.baidu.com/s/1EfffExvtxZYI1QLuxUZQ_g
提取码:5wfe
数据为plink 格式数据test.map、test.ped ;
一共包含三个品种,DOR、GMM、SUN各20个样本。
2、下载、安装detectRUNS包
install.packages("detectRUNS")
library(detectRUNS)
3、定义测试数据路径
genotypeFilePath <- ("test.ped")
mapFilePath <- ("test.map")
4、利用默认参数进行ROH检测
slidingRuns <- slidingRUNS.run(
genotypeFile = genotypeFilePath,
mapFile = mapFilePath,
windowSize = 15, threshold = 0.05,
minSNP = 20, ROHet = FALSE,
maxOppWindow = 1,
maxMissWindow = 1,
maxGap = 10^6,
minLengthBps = 250000,
minDensity = 1/10^3,
# SNP/kbps maxOppRun = NULL, maxMissRun = NULL)
以上是默认参数,实际分析需要调整,这个最关键!
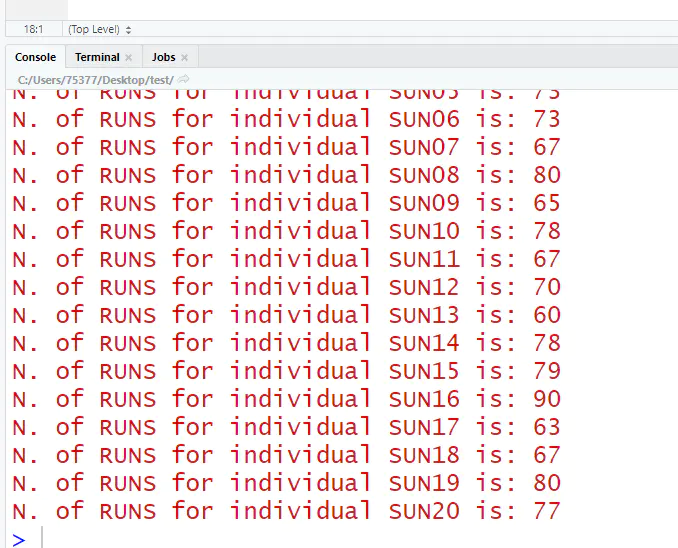
检测结果,每个个体检测的ROH数目:

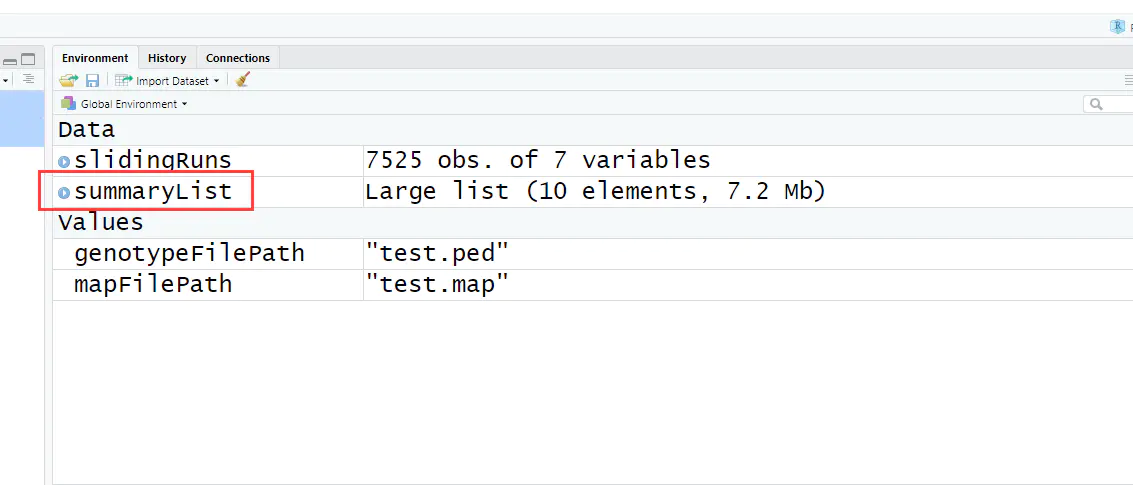
5、生成统计列表
summaryList <- summaryRuns( runs = slidingRuns, mapFile = mapFilePath, genotypeFile = genotypeFilePath, Class = 6, snpInRuns = TRUE)
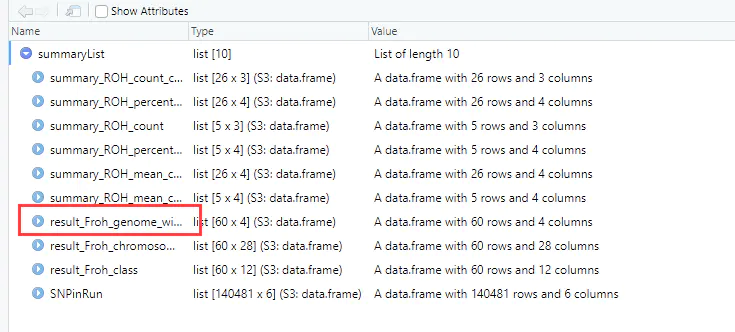
6、基于ROH近交系数如下:
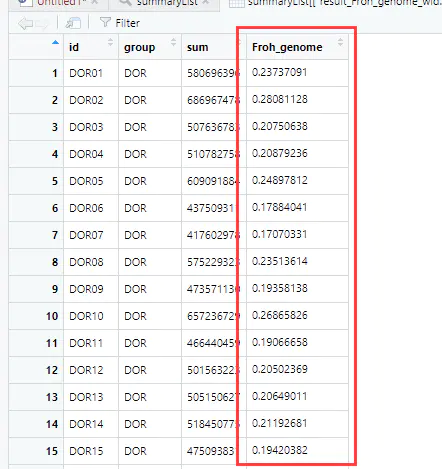
参考:http://127.0.0.1:27306/library/detectRUNS/doc/detectRUNS.vignette.html



【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· 基于Microsoft.Extensions.AI核心库实现RAG应用
· Linux系列:如何用heaptrack跟踪.NET程序的非托管内存泄露
· 开发者必知的日志记录最佳实践
· SQL Server 2025 AI相关能力初探
· Linux系列:如何用 C#调用 C方法造成内存泄露
· 震惊!C++程序真的从main开始吗?99%的程序员都答错了
· 【硬核科普】Trae如何「偷看」你的代码?零基础破解AI编程运行原理
· 单元测试从入门到精通
· 上周热点回顾(3.3-3.9)
· winform 绘制太阳,地球,月球 运作规律