文献分析 基于监督学习的细胞类型注释策略 Evaluation of some aspects in supervised cell type identification for single-cell RNAseq: classifier, feature selection, and reference construction
原文pdf连接
摘要
|
|
Progress
|
Challenge
|
Demand
|
|
Background
|
|
||
|
|
|
|
|
|
Solve
|
What
|
How
|
Effect
|
|
通过实际数据分析评估不同的策略组合
|
参考数据的影响以及参考数据的处理策略
|
提供了使用监督细胞分型方法的指南和经验法则
|
|
|
Result
|
Study design
Methods under comparison
3种现成的:random forest, SVM with linear kernel, and SVM with radial basis function kernel
2种基于scRNA相关性的方法: scmap and CHETAH
2种监督深度学习方法:multi-layer perceptron (MLP) and graph-embedded deep neural network (GEDFN)
2种半监督深度学习:ItClust with transfer-learning and MARS with meta-learning concepts
虽然还有其他方法,但基于已有的研究,SVM with rejection, scmap, and CHEAH是他们中最好的
纳入GEDFN方法是为了研究基因网络信息是否有帮助
ItClust 只是用ref数据得到非监督聚类的参考值
Feature selection methods
关键是很多基因不是类型特异的,应该去除
3中非监督变量选择:Seurat, FEAST, F-test
不选择
在ref中选择,在tar中选择
在ref中不选择,在tar中选择
在ref中选择,在tar中不选择
Datasets
人PBMC
10X,lupus patients
10X,Smart-seq2,CEL-seq2 pbmc1 fresh
10X,Smart-seq2,CEL-seq2 pbmc2 fresh
人胰腺
3个
小鼠脑
Drop-seq frontal cortex “Mouse brain FC”
Drop-seq hippocampus regions “Mouse brain HC”
10X prefrontal cortex region “Mouse brain pFC”
DroNc-seq cortex samples “Mouse brain cortex”
10X frontal cortex regions s “Mouse brain Allen”
经过分选的人PBMC数据作为标准
ref和tar来自不同的平台会怎样?
ref和tar来自不同的样本状态会怎样?
来自不同的实验室?
来自不同的组织区域?
来自不同的生理状态?
研究:整合多个数据是否提高性能?
研究:去除噪声细胞是否提升性能?
Evaluation metrics Accuracy:正确注释的在全体细胞中占比
Adjusted Rand Index:聚类相似性
Macro F1:只用于在细胞类型比例不平衡时评估精度和召回率
运行时间
Summary of the study design |
||
|
F-test on reference datasets + MLP
基于F-test的特征选择和MLP的组合 |
|||
|
Impact of the reference data size
(1) 0 to 1000 cells, (2) 1000 to 5000 cells, (3) 5000 to 10000 cells, and (4) 10,000
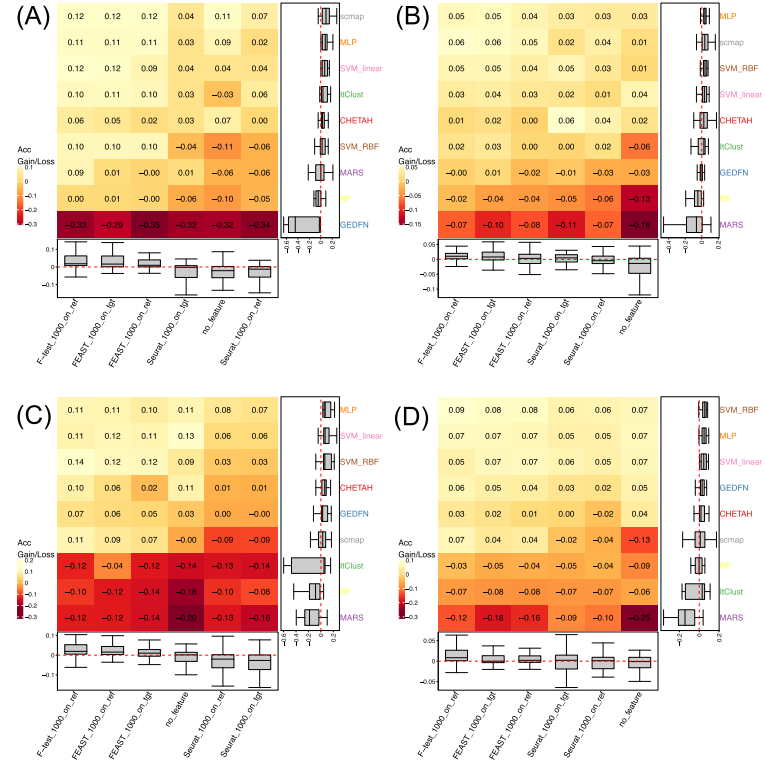 基于学习的方法细胞数越多排名越高(MLP,SVM)
|
|||
|
Impact of number of cell types
一个组织中有少数主要类型,有许多子类,子类的比较相似,不好区分
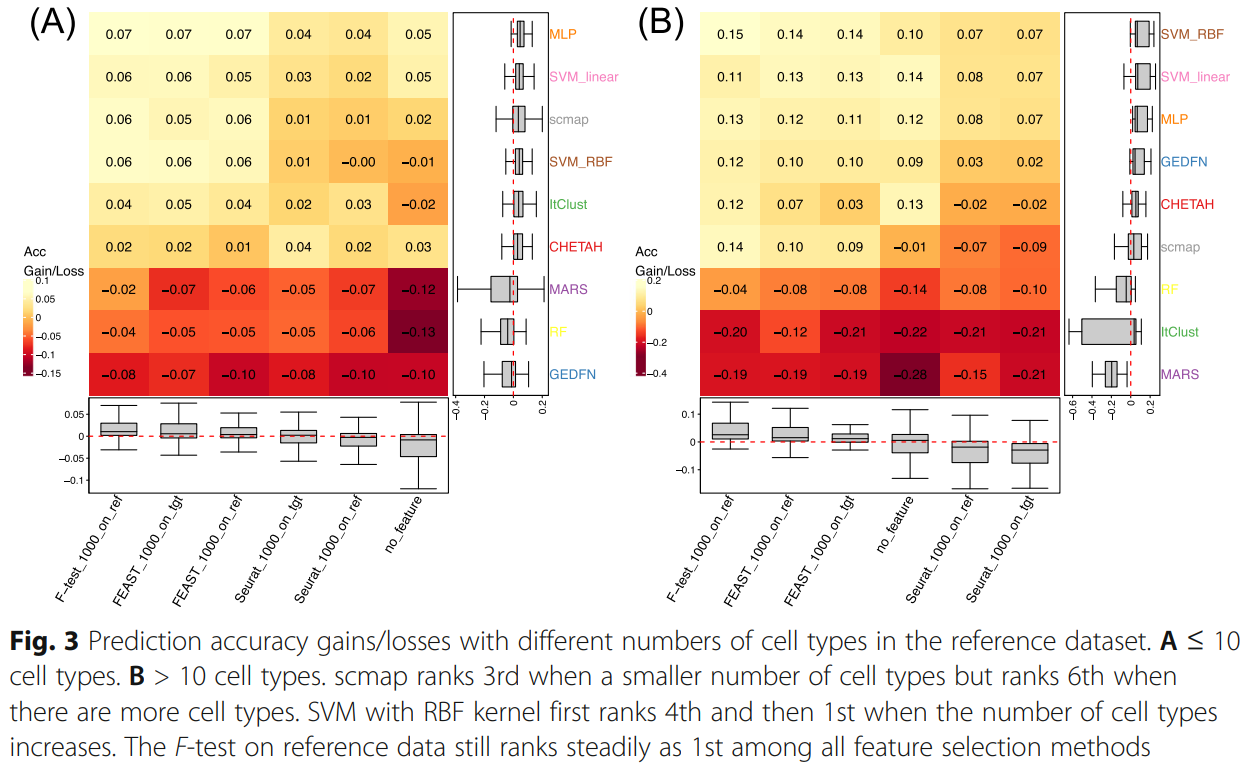 |
|||
|
Impact of cell type annotations
上述注释结果来自marker,现在以分选数据作为金标准
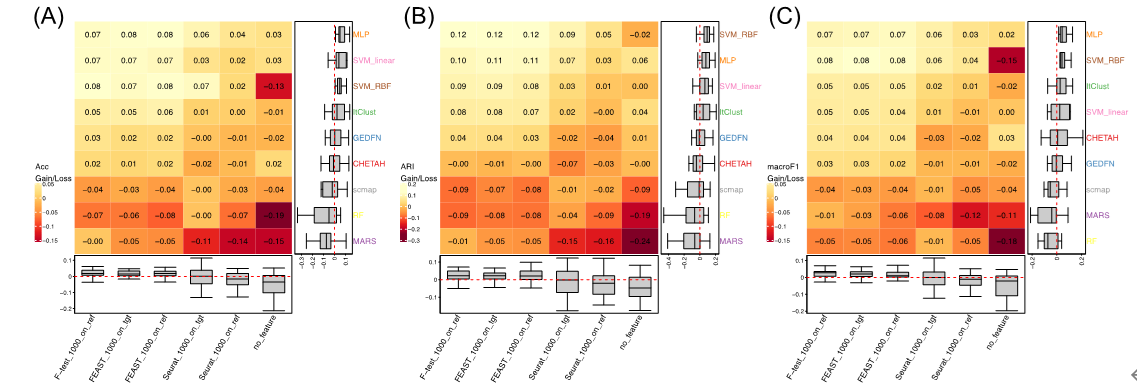 |
|||
|
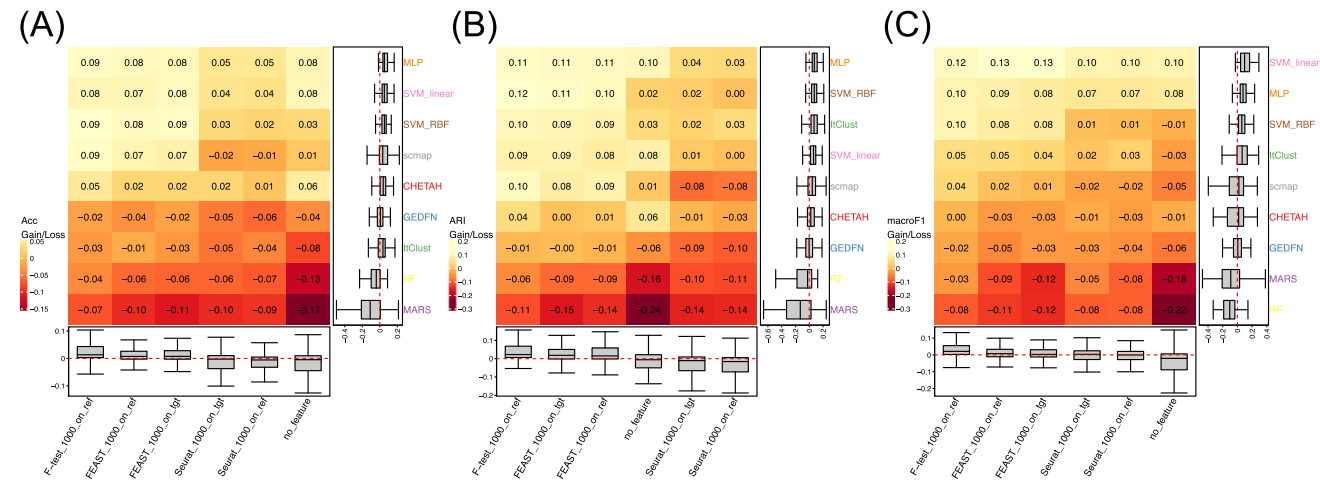
Impact of data preprocessing
评估去除batch效应或者数据插补带来的影响
先评估三种插补方法:没有明显提升,结论为不必要
在评估batch效应去除:Harmony and fastMNN(指的是ref数据和tar数据之间的batch effect)
批次效应不会影响预测性能,可能不需要校正,我们直接将数据集连接起来进行以下分析。
 |
|||
|
Drop-seq frontal cortex “Mouse brain FC”
Drop-seq hippocampus regions “Mouse brain HC”
10X prefrontal cortex region “Mouse brain pFC”
DroNc-seq cortex samples “Mouse brain cortex”
10X frontal cortex regions s “Mouse brain Allen”
Condition effect
个体差异:不同样本的差异
条件差异:技术差异
Comparing individual effect(只有样本不同), region effect, and dataset effect in mouse brain data
将“Mouse brain FC”的一个个体固定为tar数据
individual effect : 相同数据来源“Mouse brain FC”
biological effect(区域效应):“Mouse brain HC”
dataset effect:“Mouse brain cortex”和“Mouse brain pFC”
Comparing batch effect and clinical difference in Human PBMC
选择疾病的“Human PBMC lupus”作为tar数据
individual effect:同批次不同个体
batch effect:不同批次
clinical difference:不同批次不同生理状态(不同处理)
人数据差是因为包括了子类,子类多信号弱
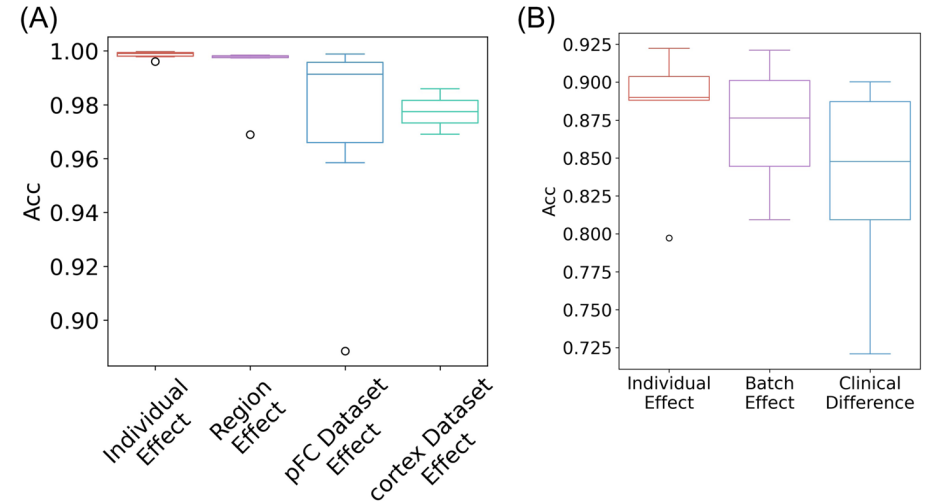 Conclusions on conditional effects between reference and target datasets
1.individual effects个体效应最小(相同条件,相同数据,不同样本)
2.biological effect对主要细胞类型的注释影响不大(不同样本,海马体的主要细胞类型可用于注释额叶皮层的主要细胞类型)
以上情况在实际中不常见,实际一般是跨数据集的
3.在跨数据集的情况下,主要细胞类型的注释效果并不受太大影响
4.在没有强烈的实验处理差异的情况下,主要细胞类型的注释不受太大的影响
|
|||
|
Pooling references improves the prediction results
固定一个数据作为tar数据
结合不同条件和不同样本的数据作为大的ref
通过抽样去除细胞数量增多带来的影响
数据集内预测:
选择“Human PBMC lupus,” 和“Mouse brain FC”数据的主要类型
选择“Mouse brain FC”的子类
数据集间预测:
用“Mouse brain FC”预测“Mouse brain pFC.”
Individual effect, pooling effect, and downsampled pooling effect
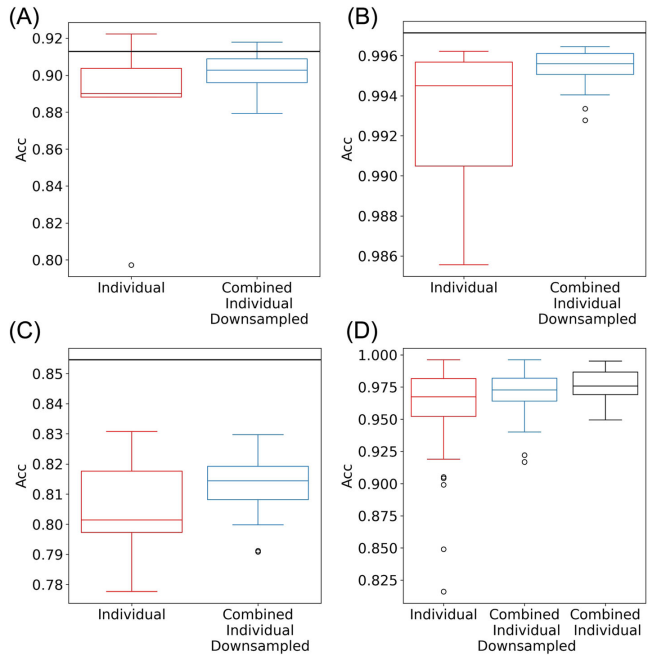 ABC是数据集内预测,蓝线是合并并抽样,黑色是合并不抽样,其中C为子类
合并对子类预测提升较大
D是数据集间预测,整合同样得到相对较好的结果
Pooling reference from different conditions can improve the prediction results
研究整合不同条件的数据带来的影响
整合不同区域和不同数据的小鼠脑数据
整合不同批次和不用处理条件的PBMC数据
首先在数据集内部整合样本,再整合数据集外部
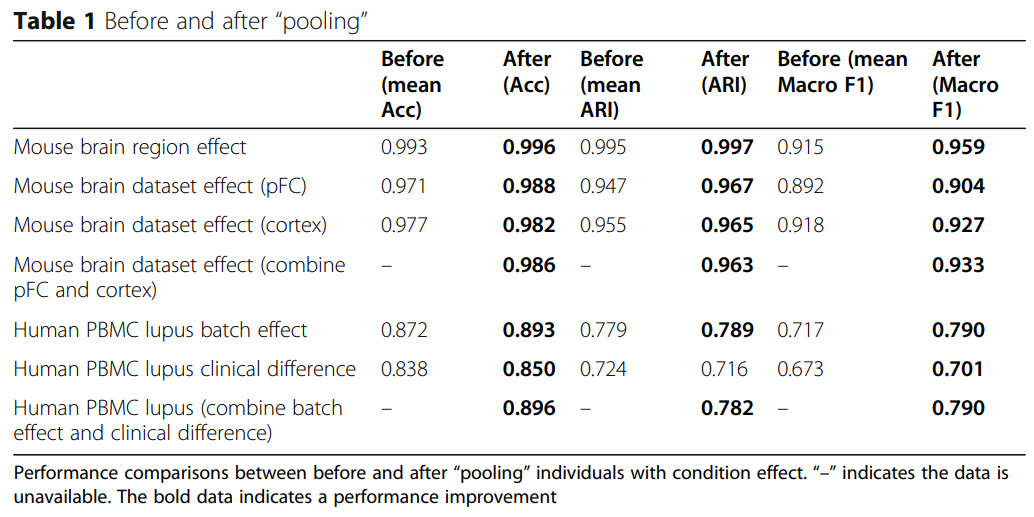 整合数据会减少不同条件带来的偏差
扩大数据集在什么时候达到饱和点?
三个方面:
(1)预测相同数据集中的主要类型
(2)预测不同数据集中的主要类型
(3)预测相同数据集中的子类
两个途径:
结合不同细胞数样本的数据作为ref
先结合数据,再随机抽样作为ref
 A是“Mouse brain FC”数据集内部的主要类型预测,细胞数增加时明显饱和
B是结合“Mouse brain pFC” and “Mouse brain Allen” 主要类型来预测“Mouse brain FC”的样本
C是“Mouse brain FC” 数据集内部的子类预测,尚未达到饱和,需要更多数据
|
|||
|
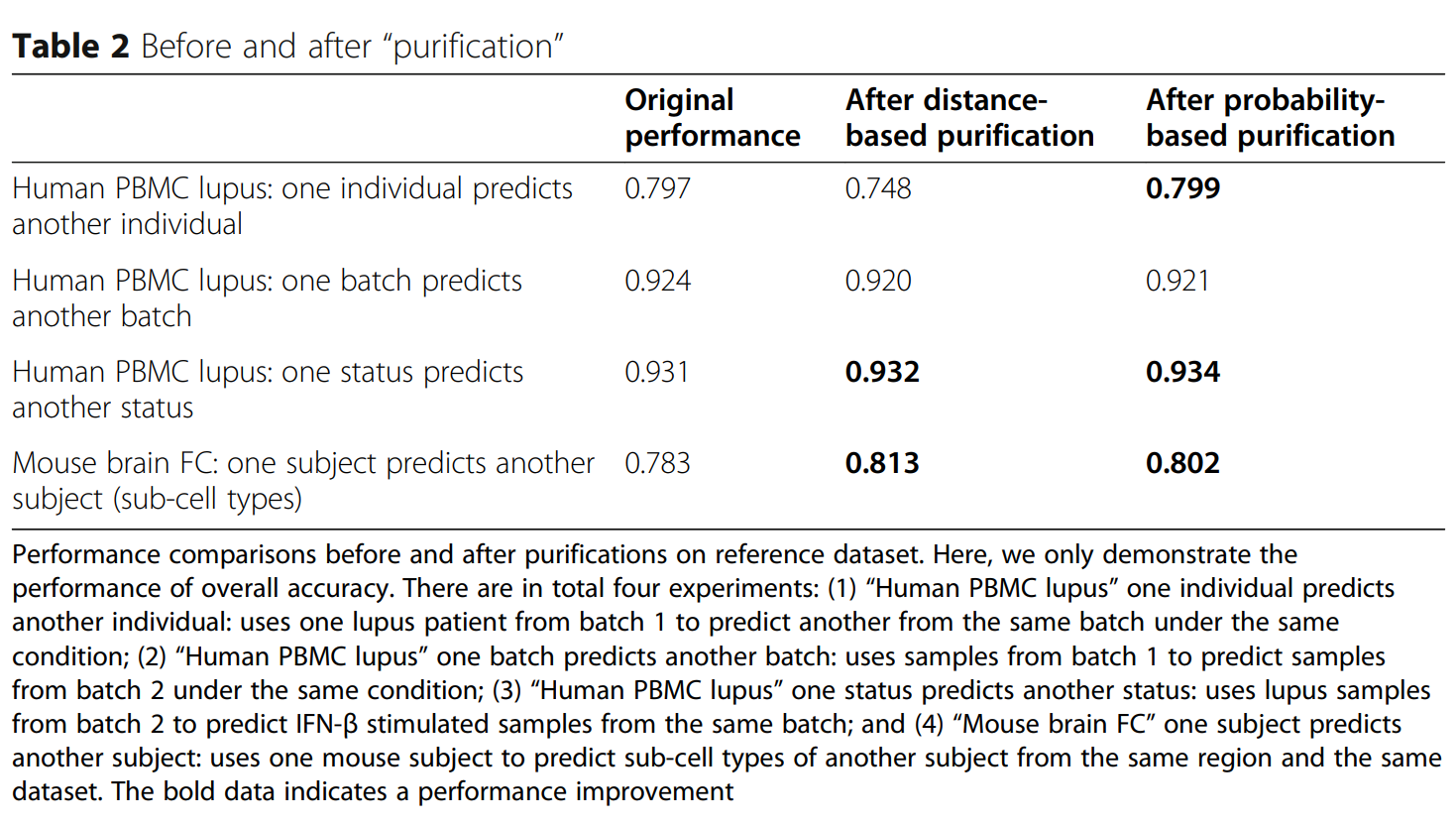
Purifying references does not improve the prediction results
两种策略:去除10%边缘的细胞
(1)基于距离的纯化
(2)基于概率纯化
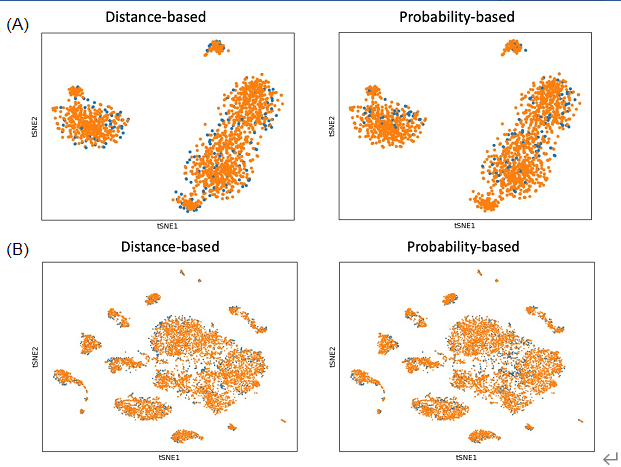 纯化对预测主要类型的提升不大,因为细胞类型的异常值对分配标签的影响不大,但是,当存在子细胞类型时,细胞簇之间的异常值会充当噪声,通过去除这些异常值,可以稍微提高预测。(是不是本质上还是数据量和信号的问题?)
 |
|||
|
Predicting sub-cell types
困难所在:
1.不同数据集的子类定义不一致
对于不同组织类型和不同条件下的数据需要做到全面+一致的细胞子类定义
如果没有这种一致的定义,我们建议:(待研究)
使用监督方法预测主要细胞类型
再对主要类型分别进行无监督注释
|
|||
|
Predicting novel cell types
监督方法只能注释已有的标签
但是可以设置打分阈值,当分数太低时将细胞注释为unassigned
移除一些ref中的细胞类型(一种或两种)
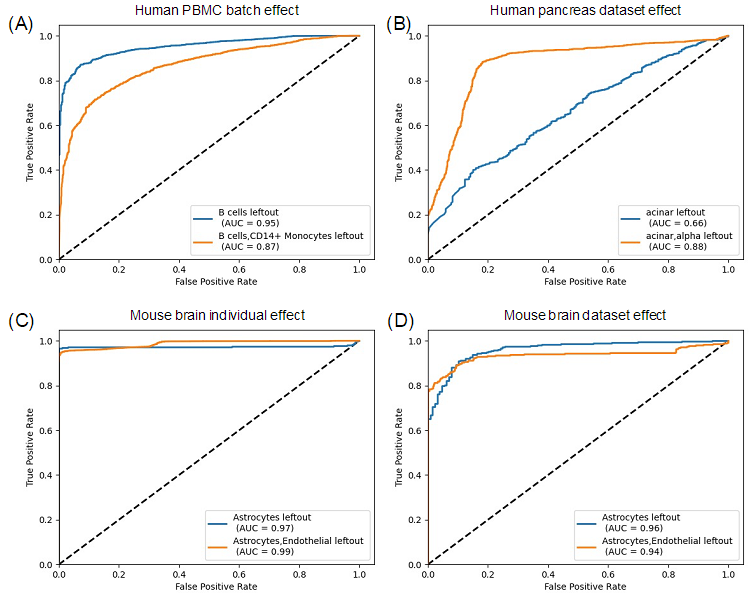 显示是否能将预测概率较低的细胞注释为unassigned
(待研究)
|
|||
|
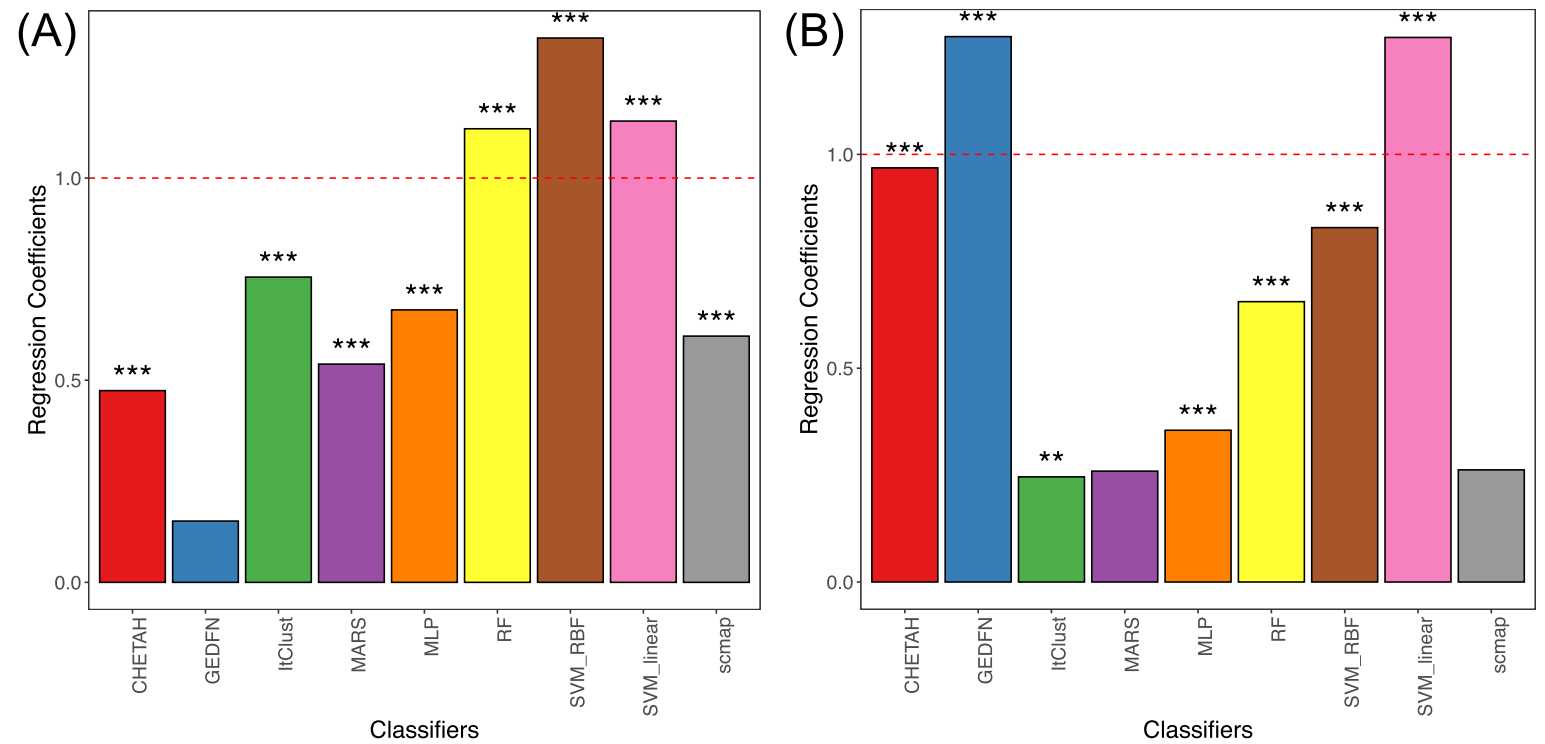
Computational performance
训练时间:ref的细胞数,ref的细胞类型
MLP方法与二者的关系较小
 |
|||
|
|
|||
|
Conclusion& Discussion
|
建议: 1.对ref数据进行基于F-test的变量选择,MLP作为分类器(ref细胞数大于5000)(SVM也行但是计算效率太低) 2.在ref细胞数小于1000时,基于相关性的scmap交优 3.ref和tar之间的差值和批次效应不用处理 4.注释是ref和tar之间的biological and clinical conditions 应该匹配,不过对主要细胞类型的影响不大 5.pooling不同数据集提升结果(关键是平均一些生物和技术的差异) 6.饱和度分析中发现加入某些数据会导致效果下降,如何选择高质量的ref数据是关键 7.纯化数据没有大用 主要细胞类型的注释比较容易,但是由于不同数据集的子类定义差异,子类注释比较困难;很可能是因为,不同biological and clinical 条件下,子类确实是不同的 所以不建议子类的直接监督注释,建议分两步: 1.监督注释完成主要细胞类型的注释 2对子类进一步非监督聚类+(marker,功能分析) |
||
|
Method
|
|
||
"The world is a fine place and worth fighting for." I agree with the second part.

 浙公网安备 33010602011771号
浙公网安备 33010602011771号