文献分析 tradeSeq Trajectory-based differential expression analysis for single-cell sequencing data
原文pdf连接
https://www.nature.com/articles/s41467-020-14766-3#Sec23
摘要
轨迹推断通过研究基因表达的动态变化,从根本上增强了单细胞 RNA-seq 研究。在轨迹推断的下游,重要的是发现(i)与轨迹中的谱系相关的基因,或(ii)谱系之间差异表达的基因,以阐明潜在的生物过程。然而,当前的数据分析进程要么无法利用轨迹推断提供的连续分辨率,要么无法查明差异表达的确切类型。我们引入了 tradeSeq,这是一个基于负二项分布的强大的广义加性模型框架,允许灵活推断谱系内和谱系间差异表达。通过结合观察级的权重,该模型还允许考虑零膨胀。我们评估了模拟数据集和基于液滴和全长protocols的真实数据集,并表明它通过对数据的清晰解释产生了生物学见解。
|
|
Progress
|
Challenge
|
Demand
|
|
background
|
scRNA-seq study cellular pathways |
data analysis challenges
|
|
|
trajectory inference (TI) methods
trajectory = lineages
a common workflow:
降维,轨迹推断
拟时间和真实时间之间的关系并不一定线性
|
轨迹differential expression (DE)方法的缺失
|
|
|
|
目前的方法,将发育轨迹离散化
|
没有利用连续的表达分辨率;
离散的比较不同cluster或者轨迹不好解释r
|
|
|
|
已有一些用于bulk RNA多时间点的分析方法
|
scRNA-seq的基因与拟时间之间的关系更加复杂
|
|
|
|
有些方法是通过将基因的表达拟合为拟时间的函数:
Monocle fitting additive models,只能处理单分支;
TSCAN12. GPfates4:a mixture of overlapping Gaussian processes,研究分支之间的差异表达;
monocle2的BEAM,研究特定分支的差异表达;
|
GPfates和Monocle 2都缺乏可解释性,因为无法指定具体差异的区域;
GPfates 只兼容二分支结构;
BEAM 仅限于 Monocle 2 软件中实现的少数降维方法,即独立分量分析 (ICA) 和 DDRTree;
|
对新方法的要求: 鉴定轨迹内部的DE,也能鉴定轨迹之间的DE; 兼容复杂的轨迹结构 |
|
|
solve
|
what
|
how
|
effect
|
|
tradeSeq
a method and software package for trajectory-based differential expression analysis for sequencing data
|
通过generalized additive models 拟合基因表达的平滑函数; 对模型参数进行具有生物学意义的假设检验; 对于0膨胀的问题,该方法使用权重,简单理解就是剔除0值; 输入为:原始表达矩阵,拟时间注释,细胞在轨迹上的权重矩阵 |
兼容所有降维和轨迹推断方法;
为不同的差异模式提供检验,提高解释性;
 通过4个真实数据比较其他方法,这些案例研究强调了tradeSeq结果的增强可解释性,从而提高了对基础生物学的理解。
|
|
|
result
|
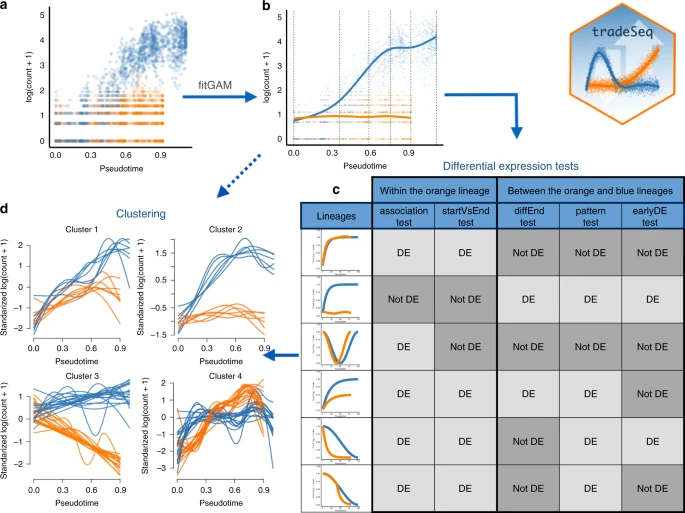
Statistical model and inference using tradeSeq通过generalized additive model (GAM)拟合
每个分支单独拟合
每个基因在每个细胞中的表达量Ygi服从负二项分布,这个分布的参数是该表达量的期望μgi以及基因特异性参数ϕg
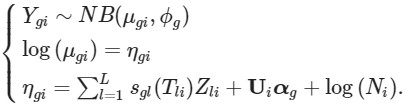 L表示不同轨迹
s表示拟合的曲线
T表示拟时间
Z表示权重矩阵
U表示细胞水平的协变量,α表示这些协变量的参数
N表示细胞之间的深度或捕获率差异
s可用K cubic basis functions的线性组合拟合:
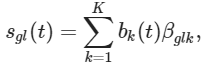 通过比较参数βglk得到轨迹内部和轨迹之间的差异基因;
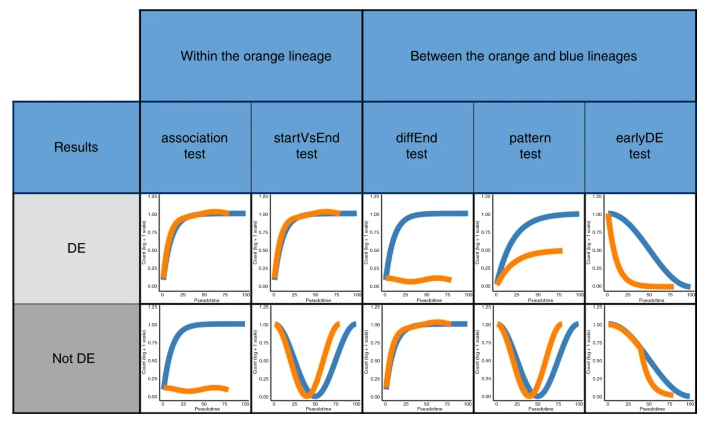
为不同的差异模式提供不同的检验策略:
 |
||
Simulation study模拟不同拓扑结构的轨迹数据:
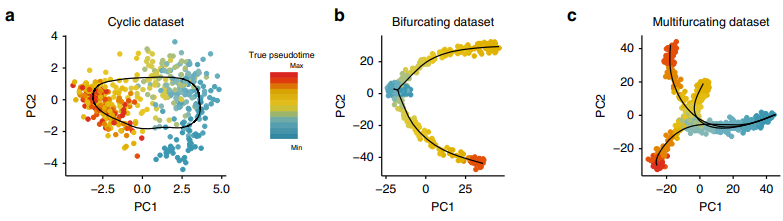 比较不同方法组合的效果:
 首先突出的是该方法兼容各种的上游方法;
然而GPfates方法甚至不能鉴定正确的轨迹,这个方法就不用考虑了;
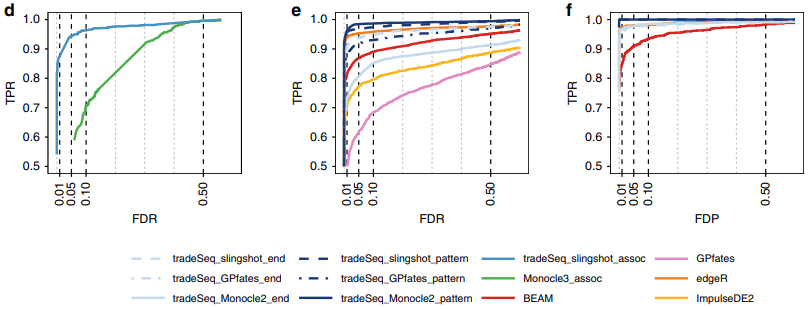
Within-lineage DE:monocle3在单分支的检验方法是Moran’s I test,tradeseq效果明显更优
Between-lineage DE:有很多方法甚至不能正确构建轨迹,在得到正确轨迹的情况下,tradeSeq效果还是很好的,tradeSeq_slingshot具有最佳性能。
总结来说;
tradeseq效果最好,
DE分析效果与上游有关,
tradeSeq_slingshot的组合效果最好。
|
|||
Computation time and memory-usage benchmark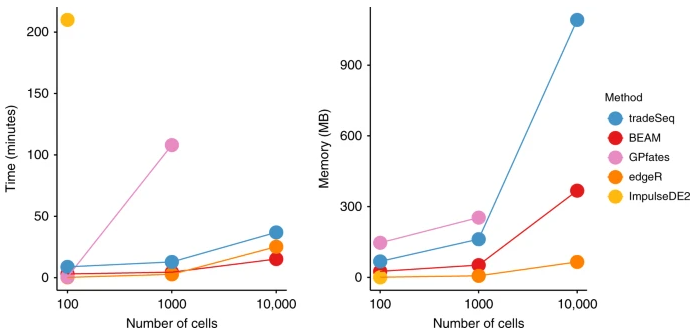 效果一般感觉,关键是代码是R写的
|
|||
Case studiesfour case study data:
a bulk RNA-seq time-course
scRNA-seq MARS-seq
Smart-Seq
10× data sets
|
|||
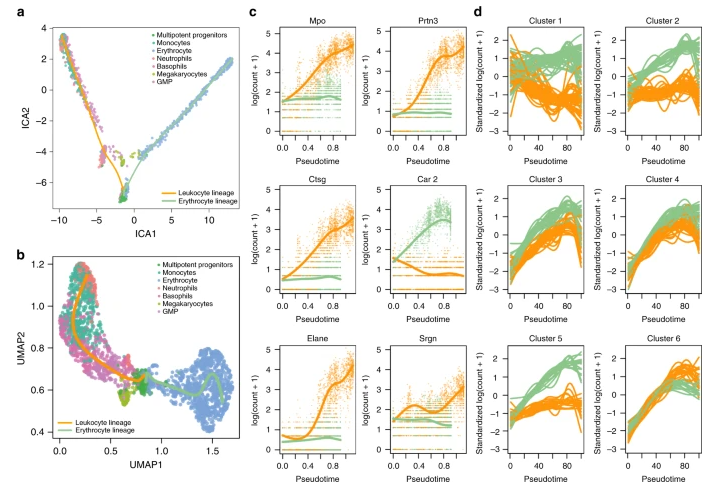
Mouse bone marrow data setDiscovering cell type markersstartVsEndTest ,diffEndTest Discovering progenitor population markerspatternTest Gene expression families基因聚类确定有相同生物学功能解释的基因
|
|||
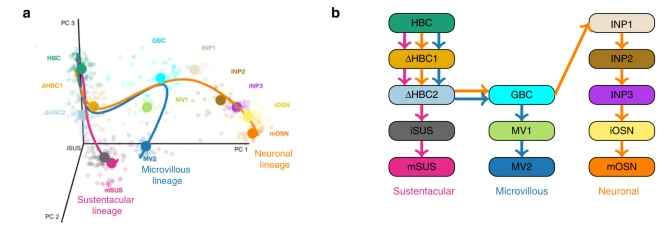
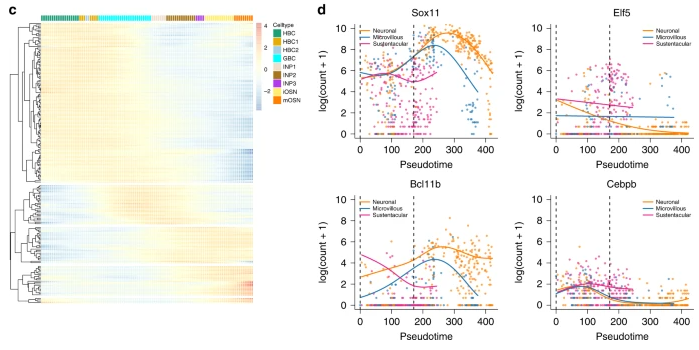
Mouse olfactory epithelium data setFletcher et al.17 study the development of horizontal basal cells (HBC) in the olfactory epithelium (OE) of mice. HBC的发育轨迹:
通过ZINB-GAMs拟合 Within-lineage DE Between-lineage DE 先前开发的用于评估谱系之间差异表达的基于轨迹的方法目前都不能适应零通货膨胀。在补充说明 5 中,我们将 ZINB-tradeSeq 分析与 ZINB-edgeR 分析进行了比较,并证明了 tradeSeq 唯一发现的基因的相关性。此外,我们说明了 earlyDETest 的功能,以识别可能驱动第一个分支点周围分化的基因
|
|||
|
Conclusion& Discussion
|
未来需要考虑拟时间的不确定性; 还需要考虑不同轨迹的拟时间是否有可比性; 展望未来,有可能通过数值最大化ZINB-GAM的可能性来一步拟合ZINB-GAMs。这可以改进我们在本文中采用的两步法,其中(i)首先使用ZINB-WaVE估计零膨胀的后验概率,然后(ii)随后用于解锁NB-GAM,以便在存在过量零的情况下进行DE分析。 |
||
|
method
|
-
|
||
"The world is a fine place and worth fighting for." I agree with the second part.

 浙公网安备 33010602011771号
浙公网安备 33010602011771号