
19-有参转录组实战5-差异基因分析
 有参转录组实战5-差异基因分析
有参转录组实战5-差异基因分析

#本教程仿自于B站的15天入门生物信息视频,若有疑惑请以视频为准。若侵请联系删除。
#新开个文件夹,DE,把gene.count.matrix文件丢进里面,编写分组文件group.txt:
WT WT1
WT WT2
WT WT3
OE OE1
OE OE2
OE OE3
#编写组比较compare.txt文件,这里是前面比后面:
OE WT
#我们后面需要用到Trinity软件包里面的一个文件,我们先下载
conda create -n Trinity#创建环境
conda install -n Trinity -c bioconda trinity#在这个环境中安装软件
#我们看看这个地址中是否有文件"~/miniconda3/envs/Trinity/opt/trinity-2.8.5/Analysis/DifferentialExpression/run_DE_analysis.pl"。
#进入R环境
source activate R
#然后输入命令:
perl ~/miniconda3/envs/Trinity/opt/trinity-2.8.5/Analysis/DifferentialExpression/run_DE_analysis.pl \ --matrix genes.counts.matrix \ --method DESeq2 \ --samples_file group.txt \ --contrasts compare.txt
#结果文件夹是DESeq2.1272686.dir,里面的OE_vs_WT.DESeq2.DE_results就是我们需要的,其它的都不要:
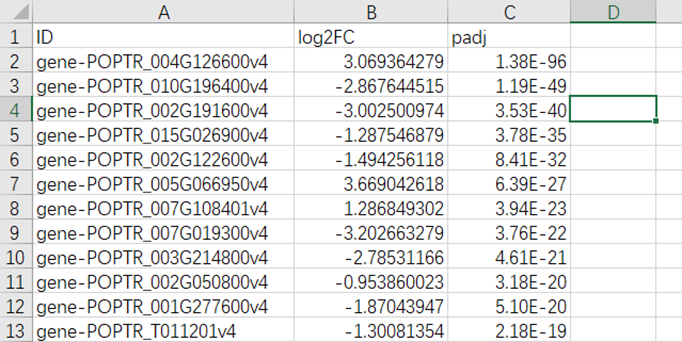
#我们打开文件后,整理成三列,如下,并将文件命名为NOT_DE.txt:
#我们需要做一个筛选条件,LOG2FoldChange>=1,padj<0.05,用一个awk命令搞:
awk 'BEGIN{OFS=FS="\t"}{if(FNR==1) print $0; else {abs_log2FC=($2<0?$2*(-1):$2);if(abs_log2FC>=1 && $3<0.05) print $0;}}' NOT_DE.txt > DE_genes_filter.txt
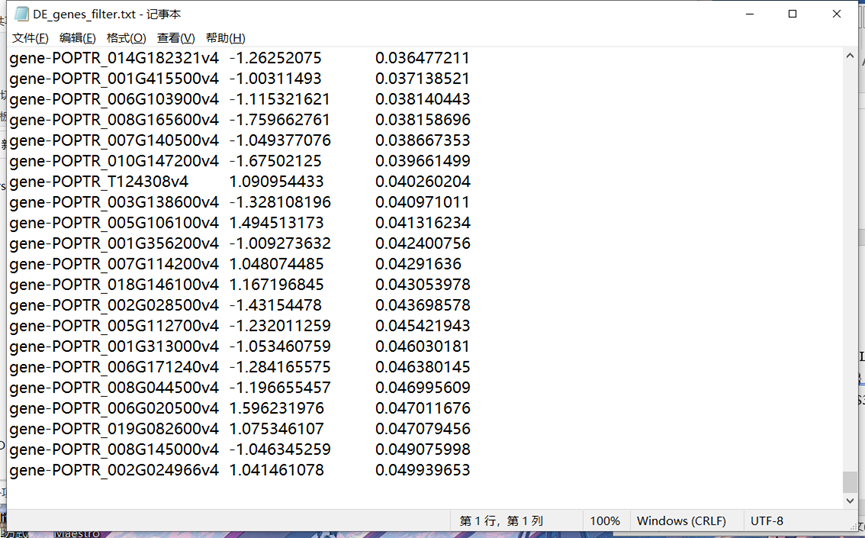
#查看最终结果,拉到最后,符合筛选结果:
#暗杀教室


【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· 震惊!C++程序真的从main开始吗?99%的程序员都答错了
· winform 绘制太阳,地球,月球 运作规律
· 【硬核科普】Trae如何「偷看」你的代码?零基础破解AI编程运行原理
· 上周热点回顾(3.3-3.9)
· 超详细:普通电脑也行Windows部署deepseek R1训练数据并当服务器共享给他人