ATAC-seq数据处理流程 | 自定义 & 个性化分析
本文的分析套路剑指10-20分的journal,全是干货。
发现一个不错的专题,讲得很详细。
其中一个细节IDR,讲得很通俗。
ATAC-seq data analysis: from FASTQ to peaks
基本概念:
- ATAC-seq和ChIP-seq鉴定出来的peak到底是一些什么区域?除了promotoer就是enhancer
- TSS enrichment和motif是两个不同的套路,TSS是特指启动区域,每个基因只有一个;另一个就是motif,这就是抓出TF,哪些TF在发挥作用
分析套路
鉴定差异peak
- 两组数据比较
- 多组数据比较 - peak cluster
处理细节:
-
Peak calling was performed using MACS2 from all sample reads. 多样本这样处理更加靠谱,macs2出来的peak结果才是靠谱的,不要随便用bedtools修改源头的peak,否则得到的peak很奇怪
-
The number of raw reads mapped to each peak at each condition was quantified using the intersectBed function in BedTools. 最终用的别人推荐的multicov
-
Raw counts in peaks were normalized using the DESeq package in R.
-
Peak intensity was defined as the log2 of the normalized counts.
bedtools multicov -bams 1.bam 2.bam 3.bam 4.bam-bed file.bed > read.count.txt
可视化差异peak
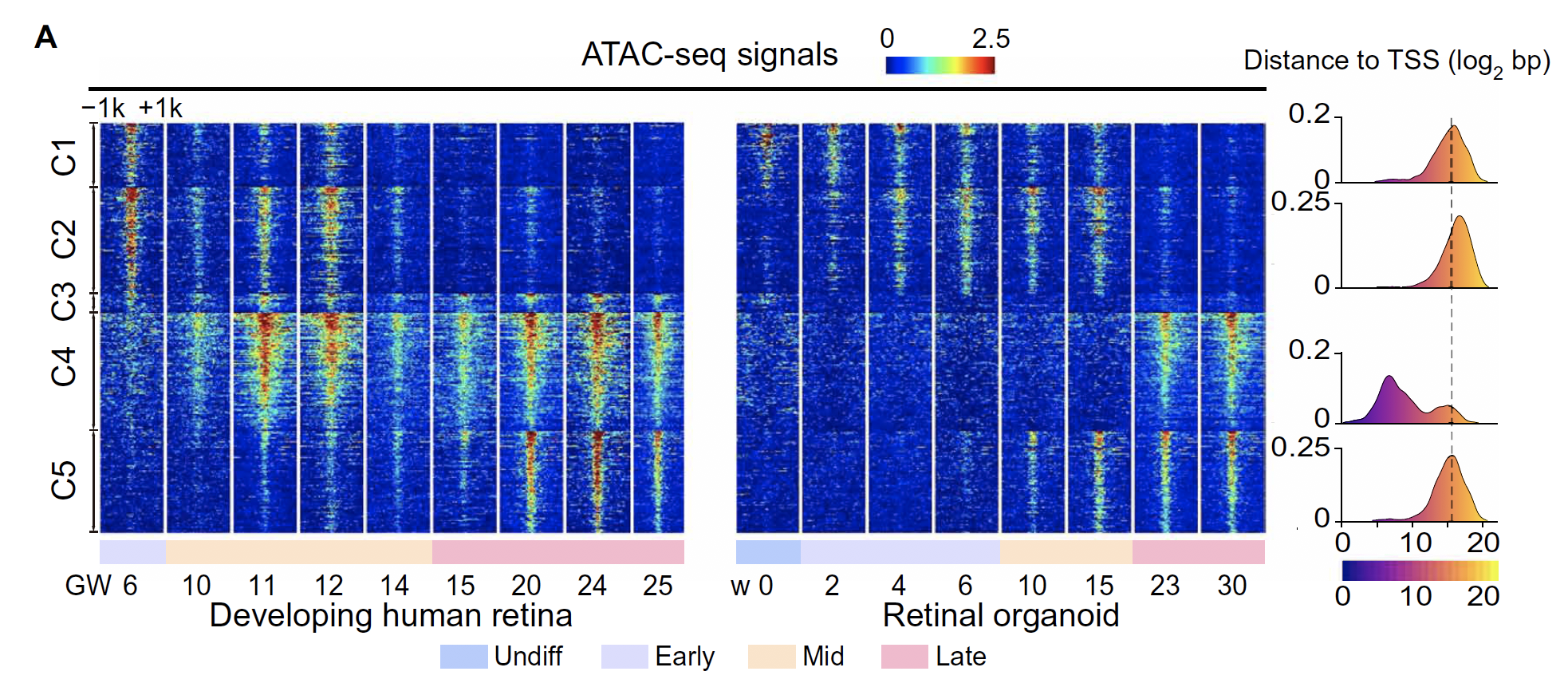
- peak signal heatmap
- distance to TSS
ngsplot这个工具比较好用,很直观,有bam就可以用,不像其他工具,一大堆复杂的衍生格式,没有搞的欲望。
ngs.plot.r -G hg19 -R tss -C example.bam/hesc.H3k4me3.1M.bam -O hesc.H3k4me3.tss -T H3K4me3 -L 3000 -FL 300
- 已经测试通了,用ngsplot来快速画图,生成数据;然后用EnrichedHeatmap来画最终的图。
- 改一下color set,能够画出媲美下面的美图了。
peak功能注释
peak邻近基因表达分析
-
expression level of genes closest to the top 1000 peaks
- 可以画二维散点图来展示ATAC-seq和RNA-seq的关系,理想情况是显著正相关关系。
motif富集分析
- TF motifs enriched in peak clusters
- 结合转录组基因表达数据验证
- 如何直接在ggplot里添加motif images,教程
- 可以直接用meme-chip一步到位
export PATH=/home/lizhixin/softwares/ATAC-seq-conda/anaconda3/bin:$PATH sortBed -g genome.txt -i up.sig.peak.bed > sorted.up.sig.peak.bed sortBed -g genome.txt -i down.sig.peak.bed > sorted.down.sig.peak.bed cut -f1-3 sorted.up.sig.peak.bed > sorted.up.sig.peak.3col.bed cut -f1-3 sorted.down.sig.peak.bed > sorted.down.sig.peak.3col.bed bedtools getfasta -fi /home/lizhixin/softwares/miniconda3/share/homer-4.10-0/.//data/genomes/hg38///genome.fa -bed sorted.up.sig.peak.3col.bed -fo sorted.up.sig.peak.3col.fasta bedtools getfasta -fi /home/lizhixin/softwares/miniconda3/share/homer-4.10-0/.//data/genomes/hg38///genome.fa -bed sorted.down.sig.peak.3col.bed -fo sorted.down.sig.peak.3col.fasta intersectBed -a /home/lizhixin/project2/analysis/ATAC-seq/encode-pipeline/ipsc/result/atac/c3097359-919e-43d3-9b81-551e4c8ff029/call-filter/shard-0/execution/glob-3bcbe4e7489c90f75e0523ac6f3a9385/IMR90-iPS_1.trim.merged.nodup.no_chrM_MT.bam -b sorted.up.sig.peak.3col.bed > tmp.bam samtools index tmp.bam ngs.plot.r -G hg19 -R tss -C tmp.bam -O IMR90-iPS_1.tss -T IMR90-iPS_1 -L 3000 -FL 300
motif印迹分析
- ATAC-seq footprint for specific motifs
- rgt-hint这个工具可以做,教程
- 搞明白什么是 TF footprint,看这篇文章:Identification of transcription factor binding sites using ATAC-seq
转录因子网络
- Cis-regulatory networks of TFs
ChIP-seq数据验证ATAC-seq结果
个别基因可视化问题 - bigwig
- UCSC genome browser看signal,都是导出pdf,自己再编辑
- IGV
- 测序数据可视化 (三) - UCSC genome browser
重点文献:
- From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis
- Chromatin accessibility analysis reveals regulatory dynamics of developing human retina and hiPSC-derived retinal organoids

 浙公网安备 33010602011771号
浙公网安备 33010602011771号