JackTalk - 知乎专栏
文章收藏
生信黑板报之RNA-Seq aligner评测
作者:popucui
链接:https://zhuanlan.zhihu.com/p/24454439
来源:知乎
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。
今天读的这篇文章来自12月12日出版的NATURE METHODS。一句简单粗暴的话总结就是,Tophat2实在弱爆了,比对错误率高,耗时长,结果还严重受参数选择影响。
文章使用的是模拟数据,分别使用人和一种叫malaria的寄生虫的数据,模拟出三种复杂度的数据集(T1、T2和T3)。对于复杂度的衡量,定义了三个尺度:
-
突变率
-
indel比例
-
错误率
T1复杂度最低,T3最高。
因为RNA-Seq测序的特性,天然的会有一部分数据延伸到内含子区,这部分跨越外显子和内含子的reads就称为『junction reads』,所以RNA-Seq比对软件需要针对此进行优化,而文章做benchmark也考虑到这点。分两个水平做比较:
-
base and read level,这点和一般的DNA aligner一样
-
junction-level
度量软件的结果时,用了两个值:
-
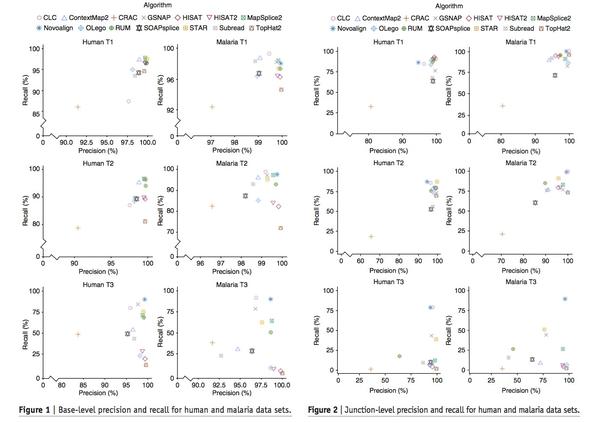
recall: 所有base中正确比对的比例。Tophat2在T1 base&read的表现,recall是85%-95%,T2降到80%,T3更是雪崩式降至20%以下。这部分表现好的是Novoalign和MapSplice2。
-
precision:所有比对上的base中,正确比对的比例。对于T1和T2,大部分软件这个值都较高。
junction-level的结论类似,不再赘述。
接下来看看调参(自行设置参数,而不是使用软件默认参数)比对,直接说结论吧,不管是否调参,表现robust的是CLC,GSNAP、Novoalign、STAR以及MapSplice2。针对复杂度高的T3数据,Tophat2调参和默认参数得到的比对率相差67%还多。
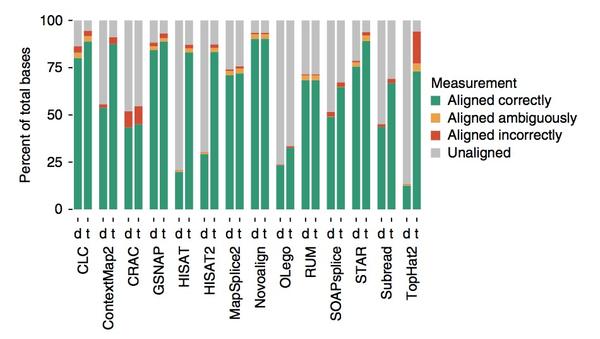
另外还有运行时间的比较,这轮表现好的是HISAT/HISAT2,其实也能看出,数据复杂度越高,比对耗时越长。
在讨论部分作者提到,随机选了20篇paper联系他们询问分析时使用的参数,13个答复的人中,10个使用的是默认错配参数(而这个参数也是最关键的),5个全部使用默认参数。最后要说的是,实际项目中使用Tophat2,通过设置一些执行规范和检查,不至于因为参数使用不当出现失误,但Tophat2的确要考虑换掉了,因为官网都已经鼓励你这么做了。
参考:simulation-based comprehensive benchmarking of RNA-seq aligners
Google 基于卷积神经网络的变异检测方法超过GATK
作者:popucui
链接:https://zhuanlan.zhihu.com/p/24475999
来源:知乎
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。
12月14日biorxiv发表了Alphabet (Google母公司) 旗下Verily生物的一个研究成果——DeepVariant,这是一种基于深度卷积神经网络的方法进行SNP和small indel检测算法,它的表现优于包括GATK在内的现有方法,而且不受测序技术和实验方法限制,也可以用于人类以外的其他物种。
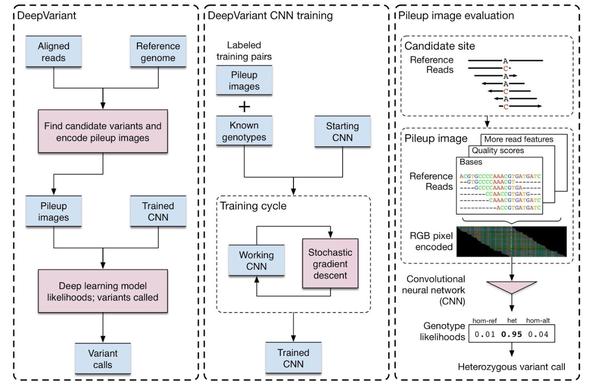
DeepVariant输入的是候选变异区域和pileup图像,将候选区域内的参考序列和支持reads支持的情况编码成一副RGB图像,而后由深度学习模型进行变异检测,最终输出的变异类型分三个并分别打分:hom-ref(与参考相同),het(杂合型),hom-alt(纯合变异)。
候选变异区域:这步主要考量候选区域支持的reads数,以及支持变异的read数与该区域总reads数之比),碱基质量值不小于10。另外仅对参考基因组上碱基是ACGT的区筛选潜在变异
介绍完原理,文章接下来拿DeepVariant和GATK分别采用两种检测变异的策略做了比较,不论是否遵从GATK best-practices,不管是反映在SNP还是indel的结果上,DeepVariant都胜出。
深度学习在图像分类上已经有了不少的成果和应用,比如安装google photos应用的手机,可以实时识别屏幕中的中文并翻译成英文。这里作者将变异检测的问题转化成图像分类的问题实在让人有眼前一亮的感觉,没想到机器学习还可以这么玩。
Verily的前身是神秘的Google X实验室下面的生命科学部,在Google对外公布Alphabet的架构之后不久毕业。
参考:Creating a universal SNP and small indel variant caller with deep neural networks
本文同步发布于知乎和公众号JackTalk
当群体变异检测遇上BWT
作者:popucui
链接:https://zhuanlan.zhihu.com/p/24515091
来源:知乎
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。
12月16日Genome Research在线发表了一篇基于Burrows-Wheeler transform (BWT)和FM-index,对2705份人类数据(包含WGBS和exome)进行SNP和indel检测的文章。作者通过和GATK、SAMtools/BCFtools进行对比,证明群体BWT的方法不仅准确可行,而且更快,随后使用所有已知病毒基因组检索群体BWT数据,发现6份样本基因组中存在人体嗜T淋巴细胞病毒的整合。
使用的2705份样本数据来自千人基因组计划,合计922 billion reads。先用Cortex做测序错误矫正,丢弃掉错误率高和质量不达标的103 billion reads。接下来做了数据截取(确保reads长度在73bp/100bp),保留正义链上的unique序列,去掉测序质量值。把reads ID和样本信息存储到一个单独数据库RocksDB (v2.6) ,以unique reads序列为键。最后用于构建BWT的reads共53 billion,合计4.9Tbp。
读完这篇文章,觉得是时候好好学一下BWT转换了。BWT在1994年被 Michael Burrows 和 David Wheeler发表,Burrows-Wheeler transform这个名字就是这么来的。
以"popucui"这个字符串为例,我们用"$"表示字符的结尾,在排序时,它的优先级也是最高的,拿"popucui$"为例,每个字符的优先级顺序是 $ > c > i > o > p > u(除"$"外,其余字符按英文字母顺序确定优先级)从左到右每次右移一个字符到"$"之后,然后对得到的矩阵按照前面讲的优先级顺序排序,就得到popucui的BWT变换结果:iuup$opc

要注意的是,每一次排序,都是以行为单位操作,每一行是一个字符串。可以看到,经过变换后相同的字符倾向于聚集到一起,这样就可以提到压缩的效率。
BWT变换是可逆的,就像文件压缩,解压之后可以得到原文件,理解这一步对学习BWT很关键。记得第一次接触BWT就是卡在这里,以至于毕业求职时,被问到Bowtie的原理,只得硬着头皮说,『先建立index』然后就扯不下去了。
还是举例子,BWT(dogwood$) = do$oodwg 这一步和上边的过程类似。接下来开始变换回原形,将尾列(就是BWT变换的结果,下面称为D)和首列合并成一个新的矩阵,然后排序(按行排序,下面的排序也是),而后将D和排序后的2列矩阵合并(D放在第一列,下面也是),再排序并将结果和D合并,如此迭代直到结果矩阵的列数为8,这时候以"$"结尾的行就是BWT变换前的字符串dogwood$
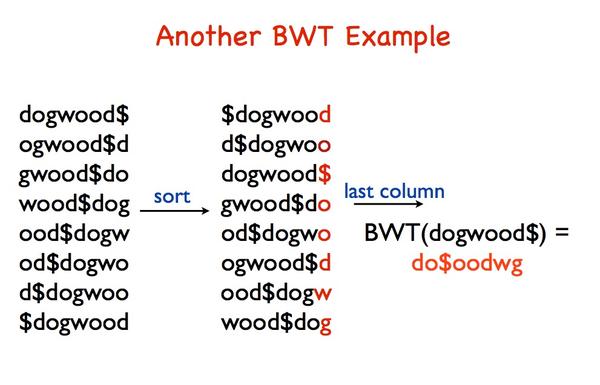
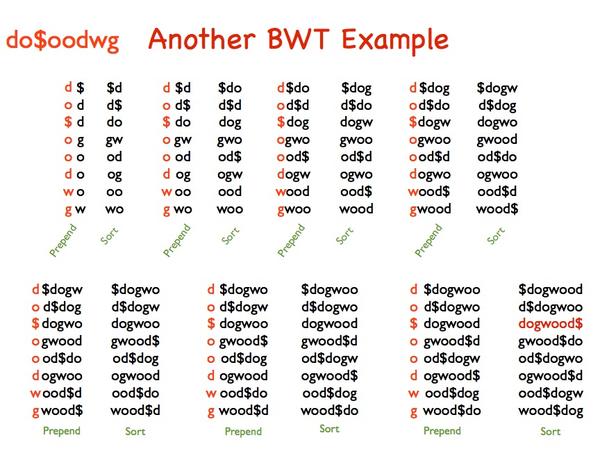 BWT最初用于文件的压缩解压,后来发现序列比对也可以采用这个算法高效进行,如今很流行的比对软件Bowtie/Bowtie2,BWA都是基于这个算法开发的。
BWT最初用于文件的压缩解压,后来发现序列比对也可以采用这个算法高效进行,如今很流行的比对软件Bowtie/Bowtie2,BWA都是基于这个算法开发的。
参考:Using reference-free compressed data structures to analyse sequencing reads from thousands of human genomes doi:10.1101/gr.211748.116
Bowtie的作者Ben Langmead写的介绍BWT的材料
生信黑板报之预见2017
作者:popucui
链接:https://zhuanlan.zhihu.com/p/24535874
来源:知乎
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。
又到了年终岁末,或是自愿,或出于公司要求,各种工作总结、performance review和个人总结集中出现。可以盘点这一年读了哪些书,做成了哪些事儿,这个世界发生了哪些变化,会影响到未来的你和我。回顾过去的意义,是为了总结得失成败,更好的面对未来。这里以个人的阅读和思考,管中窥豹,展望一下2017年的基因组学。
机器学习
AlphaGo以四胜一败战绩战胜李世石的消息,一下子让很多人惊醒一个智能时代的来临。如今机器学习正渗透到我们生活的方方面面,比如苹果的智能助理Siri,我们平时在手机上用的语音输入功能,还有锤子手机那个酷炫的『大爆炸』功能,其背后无非是图片的文字识别,而这也是用了机器学习。
除了生活中,2016年机器学习在基因组学也有很多研究成果。以biorxiv为例,用"machine learning genomics"在biorxiv搜索,得到的612条记录中,有385条都是2016年的。这些研究涉及到可变剪切,转录组注释,变异检测。预见2017年机器学习在基因组学领域的应用会更多。
增长
2016年央视的屏幕上不止一次出现测序公司的身影,再加上媒体经常爆出某某公司融资动辄千万的消息,基因检测开始走进更多人的视野。如果说2015年是基因元年的话,2017年可能是基因检测产业大幅增长的开始,这其中首先迎来爆发的,就是产前检测。
单分子测序
今年8月份nanopore tech公司发表了一篇将nanopore sequencing技术用于RNA-Seq的文章,Highly parallel direct RNA sequencing on an array of nanopores 这个技术显而易见的优势是,不用PCR扩增和反转录,直接检测核酸组成和核酸修饰形式,得到全长RNA序列。Year of the Nanopore: direct RNA-seq这篇文章的作者很肯定的说,2017年Nanopore sequencing在以下几个领域的至少一个会大显身手
细菌基因组,复杂基因组长reads辅助拼接,direct RNA-Seq
PacBio技术发表的文章今年也有不少,还是以biorxiv来看,2016年有103篇,相比之下2012-2015有61篇。PacBio的测序原理,和illumina的边合成边测序(简称"SBS")相比,在牺牲一部分准确度的情况下,得到更长的读长。看到这里,如果你还心想着『测序错误率太高,不靠谱』,那就要更新下自己的认知了。即便错误率稍高,只要错误分布随机,通过加大测序深度和错误矫正,最后可以达到很高的准确率。
前几天看到PacBio生物信息高级总监Jason Chin的一个分享Recent Progress in Long Read Genome Assembly: Theory, Practice and Future Challenges,简单统计了下,目前完全使用PacBio测序完成组装的动植物物种超过30种,包括人、羊、猪等哺乳动物。他引用一个学者对35年来de novo组装的经验概括,很有意思
错误率随机分布测序过程的取样(sampling)服从泊松分布读长足够长,可以解决重复序列问题
是的,做到以上三点,就不需要知道错误率了。
总的来说,2017年的单分子测序,会有更多的研究涌现出来,而应用于产业服务的测序平台还是illumina为主。
本文同步发布于知乎和公众号JackTalk
生信黑板报之测序错误矫正
作者:popucui
链接:https://zhuanlan.zhihu.com/p/24546764
来源:知乎
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。
测序错误只能尽可能降低,无法避免。对于illumina测序来说,伴随着反应的进行,试剂不断消耗,非同步增长(一般同样的片段,随着合成反应进行,理想情况下它们是同步增长的,这样荧光激发时,同一位置的碱基相同,发出一种颜色的荧光,这样就实现了信号放大)的片段会出现并增多,这样就会有一些点上出现杂合信号,甚至信号读成『N』。
这里介绍Heng Li的一篇博文,加上我自己的理解,聊一聊这个话题。
错误矫正用于且仅用于de novo拼接,mapping是不用的。为啥?
mapping使用的是reads全长,而错误矫正,尤其基于k-mer的算法,看的是局部信息。另外一点,mapping时使用碱基质量值,进一步提高了准确性;而de novo拼接一般没有用到碱基质量
所以,基于mapping的分析流程,不用做错误矫正。那么错误矫正的意义大吗?
事实上,对omictools上面错误矫正的软件简单统计了下,有48个之多。
长久以来,他(Heng Li)有一个看法,那就是未来有一天,我们不需要保留原始测序数据,就像我们今天不保存illumina测序的原始图像和信号强度值一样。随着越来越多人类高覆盖度测序数据的产生,最佳的保存数据的方式,不是gVCF,而是de novo组装结果,理由如下:
-
相比原始数据,组装结果更小,这点和gVCF类似
-
理想的组装结果,无损保留着原始信息,这点不同于gVCF(只记录了一些位点的基因型)
-
理想的组装结果保存了所有的变异信息,不限于SNP和short indel
-
de novo组装结果不绑定参考基因组,因而也避免了由于参考存在瑕疵引入的错误,这点不同于gVCF
-
从组装的序列出发,进行比对和变异检测可更快更好地完成
基于以上思考,Heng Li前后发表了两个错误矫正的软件fermi和BFC。
参考
生信黑板报之bioconda
作者:popucui
链接:https://zhuanlan.zhihu.com/p/24551433
来源:知乎
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。
bioconda简介
bioconda是conda上一个分发生物信息软件的频道,使用它的最大好处是,你不用自己编译软件了。
目前bioconda有超过130个添加、更新和维护生物信息软件的贡献者,他们为这个频道发布了1500多个软件包。总结起来,bioconda有以下几个特点:
-
软件是编译好的,无需自己编译
-
跨平台,支持Linux和Mac OS(本身conda还支持Windows)
-
支持多种语言,Python/Perl/R/Java/Go等
-
兼容多种语言的包管理器,如pip,CRAN,CPAN,Bioconductor,apt-get以及 homebrew
其实针对Python来说,使用conda相比pip的很大优势,就是不用自己编译。安装软件最头疼的问题,就是解决编译报错,很多时候忙活一天就为了把一个软件装好。
conda有两个版本,conda和miniconda,它们的区别是集成的软件包的数量不同:
conda集成更多常用软件包,比如numpy,pandas。而miniconda是一个瘦身版,只是集成基本功能的软件包,其余可以按需安装。
conda安装使用
conda安装很简单,到下载页面选择版本下载:
bash Anaconda3-4.2.0-MacOSX-x86_64.sh #根据版本不同,这里会有区别,3-4.2对应Python3,2-4.2对应Python2.7
conda search <package name> #查询某个软件,如果本机已经安装,会在对应版本前以“*”标示
conda install <package name> #安装软件conda install numpy=1.9.0 #指定安装特定版本
bioconda文档提到,使用之前需要先配置频道(channel),我安装的是conda,没有配置过,bioconda已经存在:
% conda config --get channels
--add channels 'defaults'
--add channels 'r'
--add channels 'bioconda'
--add channels 'https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/'
conda还有一个很赞的特性:工作环境(environment),比如我现在使用的是Python2.7,但是个别时候还需要用Python3,就可以新建一个环境:
% conda create -n py35 python=3.5 anaconda #指定使用Python3.5
% source activate py35 #切换为Python3.5
% source deactivate py35 #切换到默认版本
conda配置
conda使用的默认软件源,在目前的网络环境下很慢,尤其是安装numpy这样要下载几十M数据的情况下。是的,你可能猜到我要说什么了,换国内的镜像。在家目录下,新建一个名为“.condarc”的文件,内容参考如下:
channels:
- https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/
- bioconda
- r
- defaults
确保镜像的网址在前面,因为conda读取频道列表时是从上到下进行的。
机器学习识别cfDNA
作者:popucui
链接:https://zhuanlan.zhihu.com/p/24576148
来源:知乎
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。
最近在学习机器学习,研究了使用机器学习识别cfDNA序列特征的项目:CfdnaPattern,它使用scikit-learn实现,这里分享出来,希望给大家带来启发。
一般一个机器学习的过程可以粗略分为两步:
-
训练模型
-
选择预测变量(feature)和模型
-
数据预处理:比如要不要做 data scale,使不同变量的单位量级统一;需不需要数据降维,以消除变量之间可能存在的冗余,减少计算量
-
数据拆分:通常把数据分成训练数据和测试数据,前者用于模型训练和评估,很多时候又细分成训练数据和验证数据(validate data,确认参数调整优化后结果是否有提升);后者用于最终评估模型的表现。
-
-
使用模型:将模型用于实际数据
cfDNA(cell-free DNA,细胞游离DNA)是细胞程序性死亡而释放到血液中的一类DNA片段,通常由一类特殊物质包被(核小体),因而一般长度固定,可以用来监测人体的状态,这也是近年来大火的液体活检的研究对象。
首先是预测变量的选取,序列一般是由“ATCG”四种碱基组成,每一条序列都是如此。很自然就会想到,可以统计序列每个位点4种碱基的比例(ratio),这样就得到我们要的,可以代表序列特征的变量。作者实际上也是这样做的,统计reads上1-10位“ATCG”的比例,由于前1000条reads测序质量不稳定,如果输入reads足够,统计时跳过前1000条,如果不足,再回头统计这部分reads。这部分对应的就是选择预测变量和数据预处理。
def preprocess(options): #数据特征提取,预处理函数
data = []
samples = []
fq_files = get_arg_files()
number = 0
for fq in fq_files:
number += 1
extractor = FeatureExtractor(fq)
extractor.extract()
feature = extractor.feature() #对于reads足够多的样本,统计了1001-10000范围的reads上‘ATCG’的比例,这里是通过作者自己写的FeatureExtractor这个类实现的
if feature == None:
continue
data.append(feature) #将每一个样本的碱基比例信息保存到data列表
samples.append(fq)
训练的算法,可有多种选择,包括K近邻、SVM、随机森林等,项目自带训练好的模型,使用的是第一种。使用的训练数据有:
-
阳性: 真实的cfDNA序列
-
阴性:基因组DNA序列和FFPE样本测序数据
以上数据在文件名中分别标明(如cfdna/ffpe/gDNA)。训练完成后,使用Python的pickle模块把结果保存,以备以后预测使用。
结果的准确度,可能这也是大家很关心的问题,当然我也不例外。我从SRA随便挑了一个cfDNA的样品,SRX1591729测试结果是符合的,除了预测结果,还会画碱基比例分布图,如果同时做了非cfDNA样本的预测,放在一起看会发现明显不同的分布模式。
更多准确度的信息,软件的作者没有给出,这里凭我自己的理解给出提高准确度的一些方向:
-
采用更多的训练数据,把对reads的扫描范围延长(比如0-30)
-
预测时增加统计的reads数(比如2w-5w)
沿着这个思路,其实还可以做更多有意思的分类,比如甲基化和RNA,动植物和微生物可能都有独特的碱基分布模式,但是也要注意,不同测序平台可能对碱基分布模式也有一定影响。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号