
宏基因组数据处理方法
数据下载
wget下载
宏基因组的数据主要分布在两个数据库:1. NCBI的SRA数据库,2. ENA。近年来也有许多研究者将数据上传到中国的数据库:NGDC
你可以直接通过网页下载数据,或者是通过各个网站提供的下载工具进行批量下载。也可以到 sra-exporter 这个网站上输入项目号获得样本的下载链接。用wget或者其他下载工具进行下载,示例的命令如下:
wget -c ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR185/004/ERR1857004/ERR1857004_1.fastq.gz
wget -c ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR185/004/ERR1857004/ERR1857004_2.fastq.gz
这样我们就下载了ERR1857004样本的双端测序数据。
Aspera下载
除了wget下载以外,也可以通过安装 Aspera 来进行加速下载,
# 建立安装目录
mkdir ~/app/aspera
cd ~/app/aspera
# 下载aspera软件
wget https://download.asperasoft.com/download/sw/connect/3.6.2/aspera-connect-3.6.2.117442-linux-64.tar.gz
tar -zvxf aspera-connect-3.6.2.117442-linux-64.tar.gz
# 安装aspera
sh aspera-connect-3.6.2.117442-linux-64.sh
mv ~/.aspera ~/app/aspera
# 测试Aspera能否使用
~/app/aspera/.aspera/connect/bin/ascp -h
# 设置环境变量
echo 'export PATH=~/app/aspera/.aspera/connect/bin/:$PAHT' >> ~/.bashrc
source ~/.bashrc
ascp -h
若链接失效,点击这里 下载我的备份,或者使用conda下载
# 安装aspera
conda install -c hcc aspera-cli
ascp -h
成功安装aspera后,检查下安装目录下是否有 asperaweb_id_dsa.openssh 文件,一般在 .aspera/connect/etc/asperaweb_id_dsa.openssh
同样是到 sra-exporter 上获取aspera下载链接,这个网站国内访问可能会有一些问题。
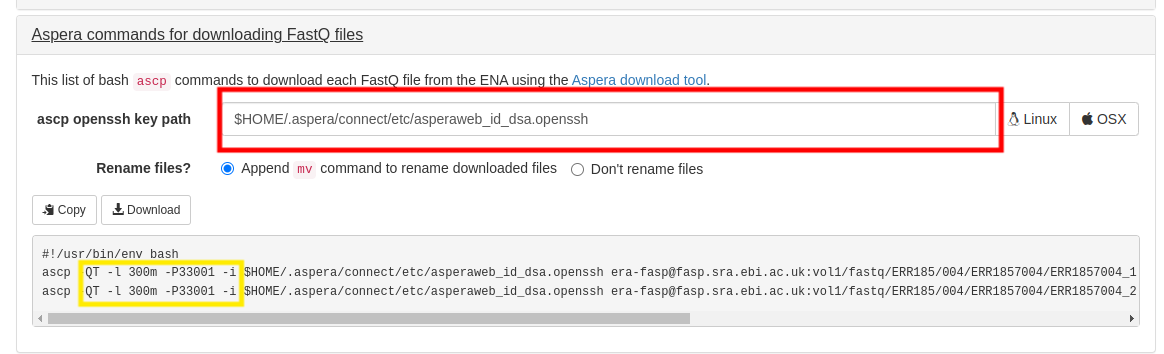
红框中填写你的asperaweb_id_dsa.openssh的路径。
黄框中没有断点续传的参数很危险,建议加上 -k1,如何加的问题——用正则,不演示了。
质量控制
FastQC安装
在进行质量控制之前需要查看质量报告,质量报告可以使用FastQC软件生成
# 安装fastqc
cd [pathway/of/fastqc/]
wget -c http://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.12.1.zip
unzip fastqc_v0.12.1.zip
# 设置执行权限
cd FastQC
chmod u+x fastqc
# 配置环境变量
vim ~/.bashrc
export PATH="/pathway/of/fastqc/FastQC:$PATH" # 添加至文件最后一行
source ~/.bashrc # 使配置文件生效
fastqc --help # 测试
同样的备份链接 在这里 或者使用conda进行安装
conda install -c bioconda fastqc
fastqc --help
fastqc只是一个观看质控情况的软件,需要根据fastqc的结果进行具体的质控,而具体的质控工作是由指控软件来完成的,常见的质控软件有 fastp ,trimmomatic。本文以 fastp 为例,在安装fastqc的同时请把fastp也安装了
conda install -c bioconda fastp
FastQC的使用及报告详解
fastqc [fastq] -o [out_file] -f [file_type]
# 当然也可以多个文件同时进行质控
fastqc [fastq_1] [fastq_2] ... [fastq_n] -o [out_file] -f [file_type]
常用参数说明
-o --outdir FastQC生成的报告文件的储存路径,生成的报告的文件名是根据输入来定的
-f --format 指定输入文件的格式
--extract 生成的报告默认会打包成1个压缩文件,使用这个参数是让程序不打包
-t --threads 选择程序运行的线程数,每个线程会占用250MB内存,越多越快咯
--min_length 设置序列的最小长度,≥最长read的长度
-c --contaminants 污染物选项,输入的是一个文件,格式是Name [Tab] Sequence,里面是可能的污染序列,如果有这个选项,FastQC会在计算时候评估污染的情况,并在统计的时候进行分析,一般用不到
-a --adapters 也是输入一个文件,文件的格式Name [Tab] Sequence,储存的是测序的adpater序列信息,如果不输入,目前版本的FastQC就按照通用引物来评估序列时候有adapter的残留
-q --quiet 安静运行模式,一般不选这个选项的时候,程序会实时报告运行的状况
这里是官网提供的一个坏的质控例子 >点击此处查看<
fastqc的质控报告一共有11个模块
Basic Statistics
这个模块是不会报错的,包含了文件名, 文件类型,处理的序列总数,序列长度,GC含量等基本信息

Per base sequence quality
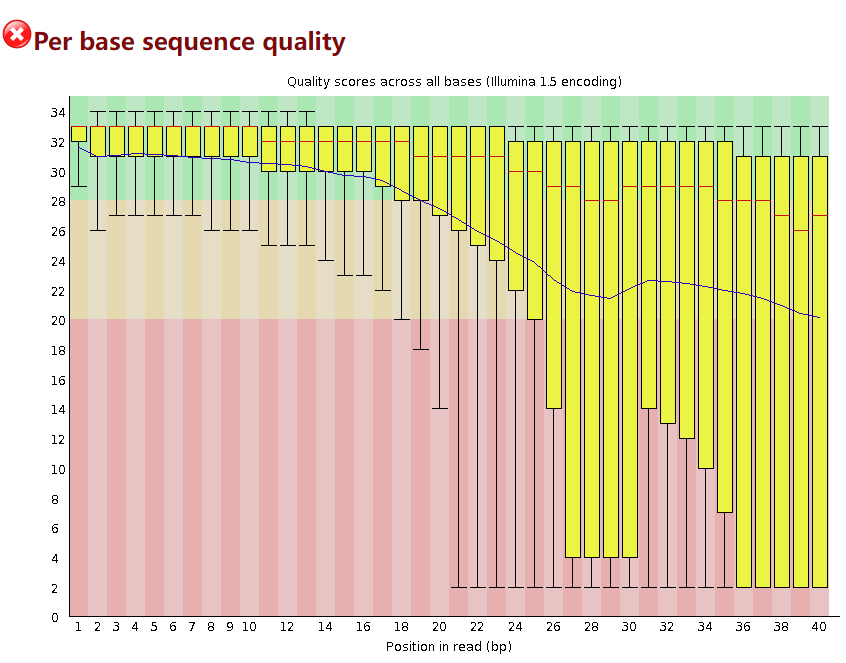
这个模块表示每个位置碱基的整体质量如何,横轴表示read上碱基的位置(第几个碱基),纵轴表示质量分数,是以Q值来展示的,Q30表示可信度为 99.9% ,Q20表示可信度为99%,Q10表示可信度为90%。
一般这个模块报错的修整方法是通过剪切掉低质量的碱基。
# 单端
fastp -i [seq] -o [outseq] -5 -W 4 -M 20
# 双端
fastp -i [seq_1] -I [seq_2] -o [outseq_1] -O [outseq_2] -5 -W 4 -M 20
这个参数的意思是从5端开始以4bp碱基为窗口进行滑动,删除质量低于Q20的碱基,双端的以 -i 参数的5'端开始
Per Tile Sequence Quality
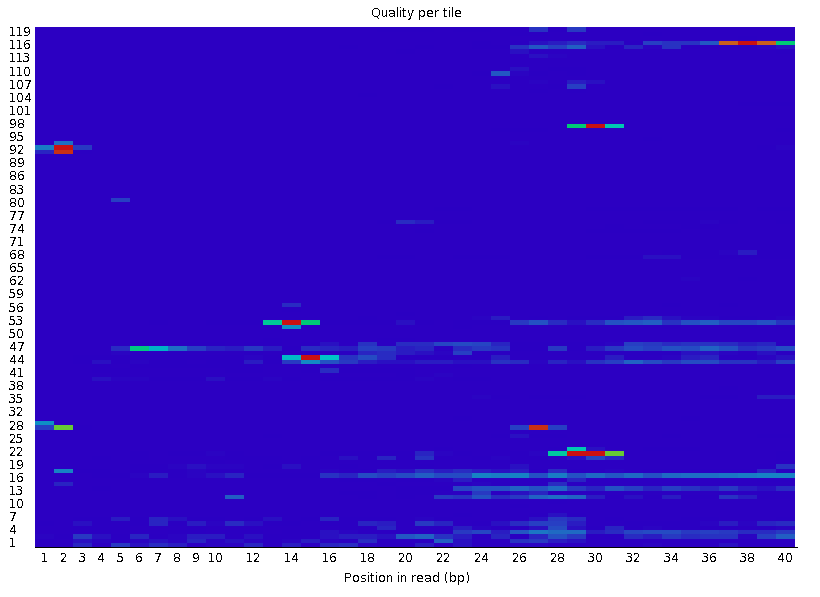
这个模块中横轴代表的是碱基的位置,纵轴代表的是tile的index编号,tile表示的是高通量测序仪上的一小块矩形区域,冷色代表在平均质量附近,暖色代表比平均质量低,正常来讲需要去除掉质量比较的低的部分tile,但是fastp中没有针对于tile编号进行去除的参数,其他质控软件中我也没有找到,所以这一部分质控如何质控目前不清楚,我认为如果上一步, Per base sequence quality的质控做到位了,所有的碱基都在Q20甚至Q30以上,这一部分质控应该是可以省略的,因为这个质控部分展示的是低于平均质量tile,但是平均质量本身就很高(Q30)那么即使低一些也能达到Q20的标准。
Per sequence quality scores
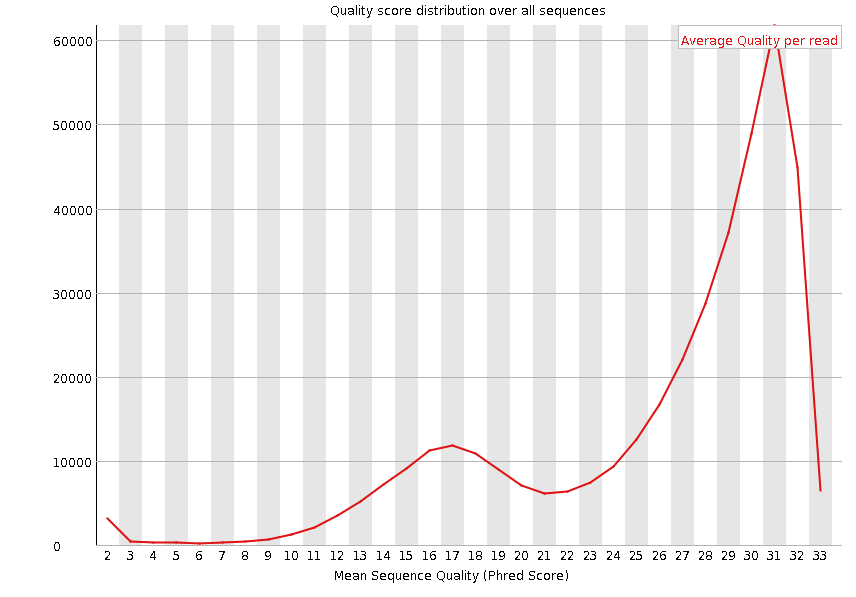
这个模块是展示每个read平均碱基质量的”频率图“,横轴代表的是read的平均碱基质量,纵轴代表read数量
同样的如果第一步的质控到位了,这步也不会有太大问题,一般来说不需要过多关注。
fastp的默认参数会过滤掉平均碱基质量低于15的read。
# 单端
fastp -i [seq] -o [outseq] -q 15
# 双端
fastp -i [seq_1] -I [seq_2] -o [outseq_1] -O [outseq_2] -q 15
如果这一项已经过关,或不想进行这项的过滤则 将 -q 参数替换为 -Q 即可
Per base sequence content
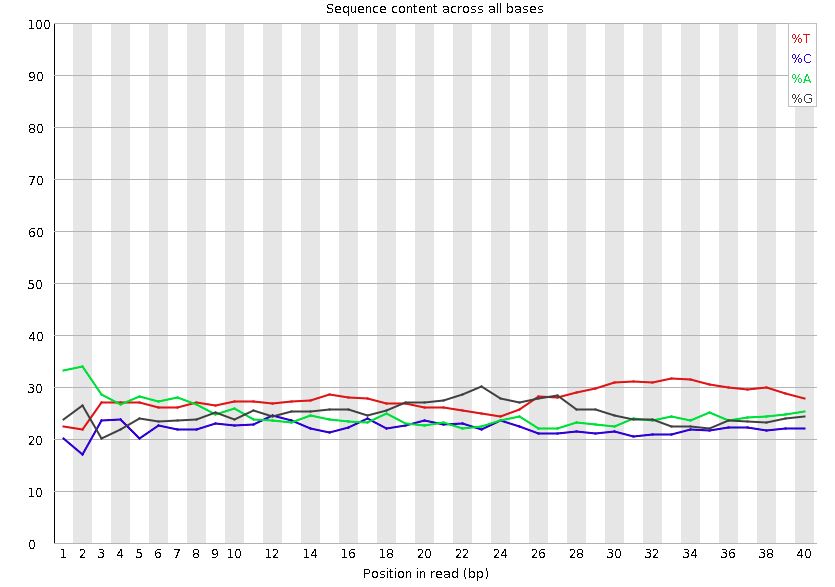
这张图片展示了每个碱基位置的ATGC的比例,正常来讲这四条线不会有太大波动,因趋于平行,且 $G \approx C, A\approx T $,如果出现了波动可能有以下原因:1. 由于在构造文库的时候,引物对DNA片段开头的碱基有一定的选择性会导致开头的碱基含量出现紊乱现象。2. 由于测序仪调整造成前几个测序结果不准确。事实上大部分情况都是由原因1引起的,如果是原因1引起的紊乱现象大部分情况不会对下游分析产生影响[3] 。但是同样的,如果你的序列足够长,切掉开头的一小部分对你序列整体的影响很小,那么我认为还是去除开头的这一部分比较好。下面是fastp一刀切去除开头序列的参数。这一部分消耗的计算资源很少。
# 单端
fastp -i [seq] -o [outseq] -f [cut_num] -t [cut_num]
# 双端
fastp -i [seq_1] -I [seq_2] -o [outseq_1] -O [outseq_2] -f [cut_num] -t [cut_num]
-f : 从 read1 的开头开始剪切 [cut_num] 个碱基,当双端测序选择这个参数时也同时会剪切read2 开头的碱基,如果想要双端的两个文件剪切不同的值则,加 -F 参数指定read2开头剪切的数量
-t :从 read1 的尾部开始剪切 [cut_num] 个碱基,同样的也可以用 -T 指定 read2 尾端
Per sequence GC content
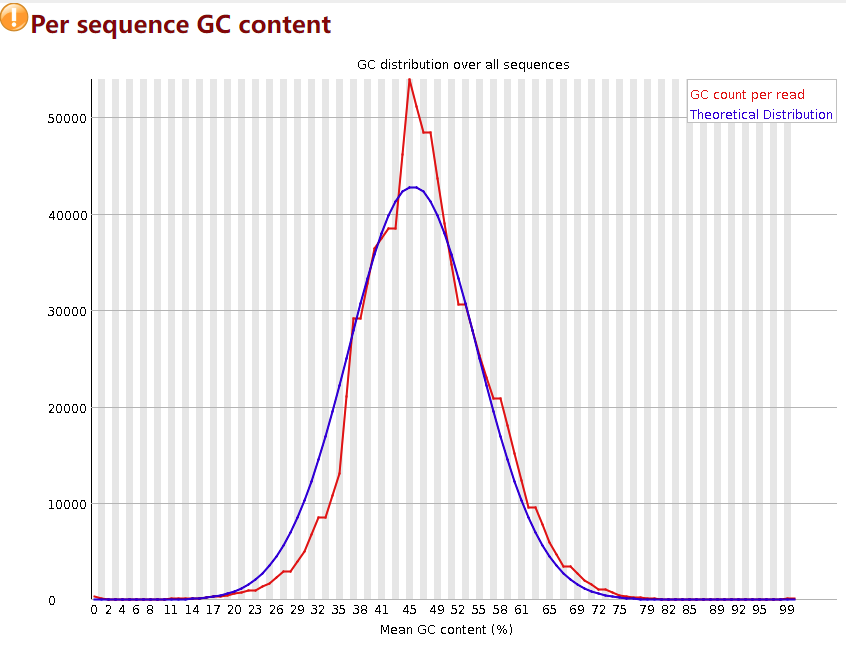
然后是每个序列的GC含量,横轴表示GC含量的平均值,纵轴表示read数量,蓝色线是标准线,是原始数据没有被污染的理想分布情况,即符合正态分布。红线是实际情况,偏差15%报warning,偏差30%报fail。如果红线,也就是实际的GC含量分布图偏离了正态,有以下几种原因:1. 意味着存在污染物,如果是宏基因组数据那么意味着很大概率存在宿主污染。2. 某种序列过度表达,影响了整体的GC含量分布。
针对于宿主污染,fastp无法进行解决,因为宿主污染的去除需要依靠数据库比对,fastp不具有这种功能,bowtie2和kneaddata等软件可以用于宿主去污染。如果是重复序列的原因,下面会有介绍如何通过fastp去除重复序列,这里先按下不表。
Per base N content
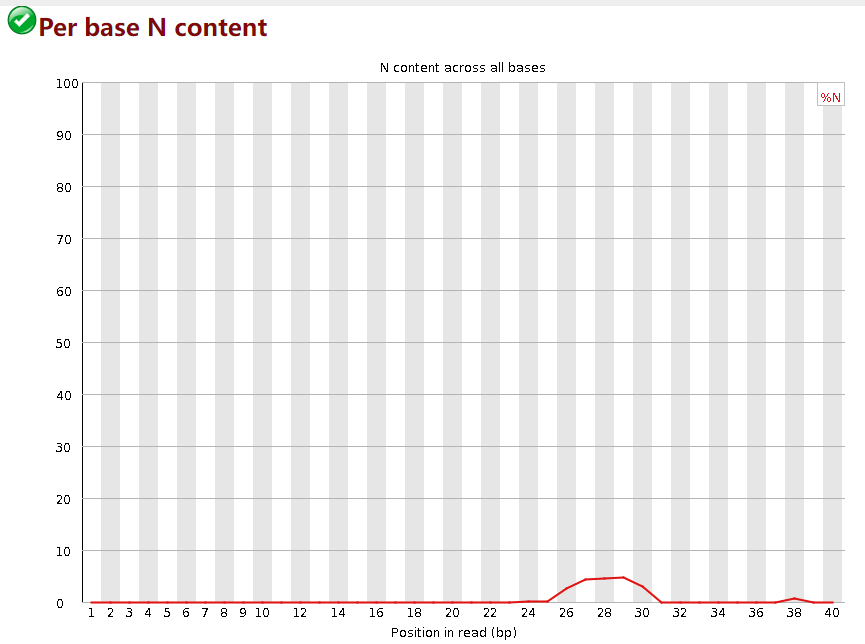
这张图表示序列的”N碱基“含量,所谓”N碱基“就是测序仪无法准确判断某个碱基时用N来替代ATGC,表示未知。通常N含量不会太高。图中,横轴是碱基位置,纵轴是N碱基的数量。当任意碱基位置的N含量>5%时报warning,>20%时报fail。
这个质量控制fastp的默认参数会做掉,一般不需要调,参数是 -n/--n_base_limit 默认值是5,即每个read若N碱基含量>5%,则这个read被丢弃。
Sequence Length Distribution
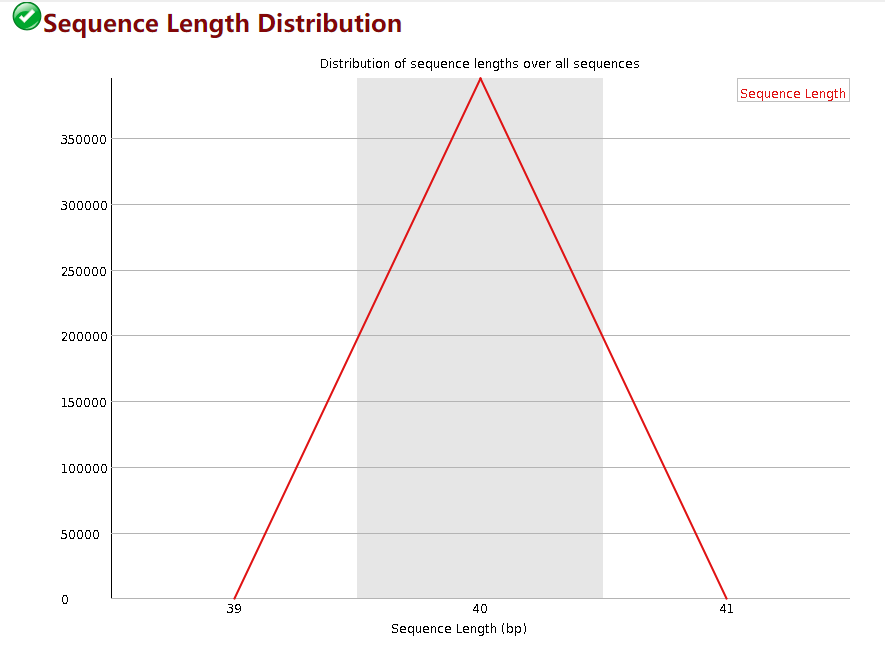
这个模块展示的每个read的长度,横轴是read长度的区间,纵轴是read的数量。一般来讲这个质控不需要去管他,因为即使read存在一些不整齐也不影响后续的比对。fastp的默认参数是去除长度低于15bp的短read。你可以通过-L 参数关闭长度过滤,也可以通过 -l 参数指定过滤的长度。
Sequence Duplication Levels
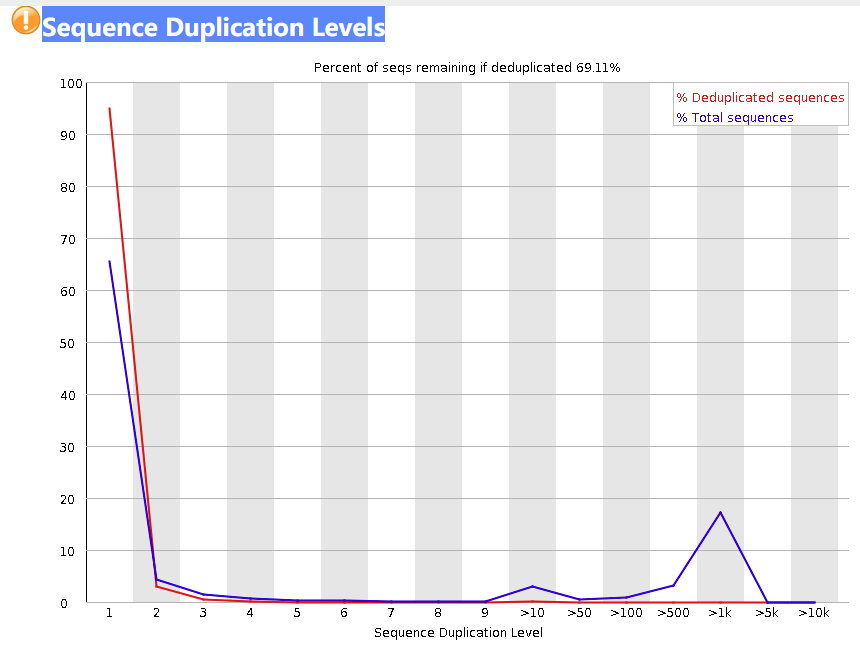
这张图表示了不同重复的read占比,横轴是read重复的次数,纵轴是read的百分比。红线和蓝线的计算方式不同,总的来说都是反应read的重复情况。蓝线的百分比是相对于总读数计算的,红线的百分比是相对于不同序列的总数计算的。
下面是一个具体的例子,思路来自于这篇文章。这里提供一个新的案例以供理解
案例:20 个 独有的read + 3 个 read (每个read出现2次) + 2个 read(每个read出现3次)
\(duplication = 1时: 蓝线_{d1} = \frac{20\times1}{20+3\times2+2\times3} = 0.625, 红线_{d1} = \frac{20}{20+3+2} = 0.8\)
\(duplication = 2时:蓝线_{d2} = \frac{3\times2}{20+3\times2+2\times3}=0.1875, 红线_{d2}=\frac{3}{20+3+2}=0.12\)
\(duplication =3 时:蓝线_{d3} =\frac{2\times3}{20+3\times2+2\times3}=0.1875, 红线_{d3}=\frac{2}{20+3+2}=0.08\)
且无论是红线还是蓝线折现下面积均为1。
也有人认为转录组数据这个部分偏高是没有关系的[4],从原理上讲,转录组数据的文库是由mRNA反转录的cDNA构成的,而mRNA有大量重复的输入是正常的。
如果你想进行这一部分的质控,也就是去除过多的重复序列,那么可以用 -D参数,
# 单端
fastp -i [seq] -o [out_seq] -D
# 双端
fastp -i [seq_1] -I [seq_2] -o [out_seq_1] -O [out_seq_2] -D
--dup_calc_accuracy 可以设置精度,1~6,1最低,6最高。
Overrepresented sequences
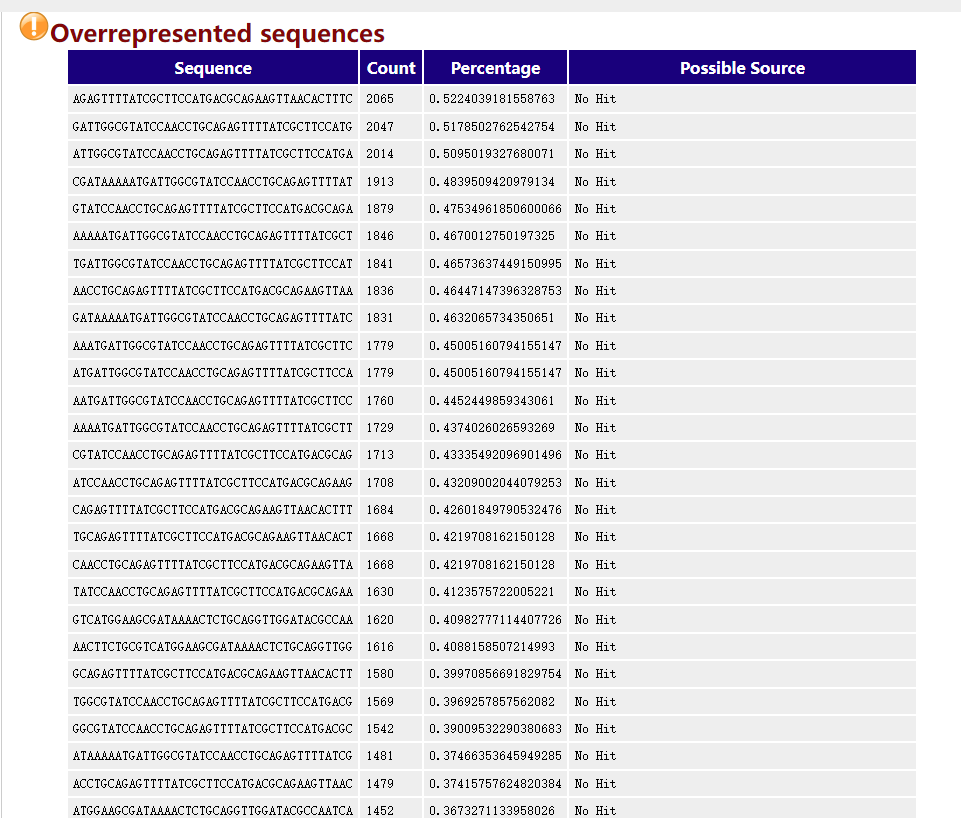
这个模块表示某一个read或者某几个read的含量过高,可能表示这几个read具有高度生物学意义,或者是文库受到了污染,fastp默认是不启用过滤的,-p 参数开启过量表达测序。
Adapter Content
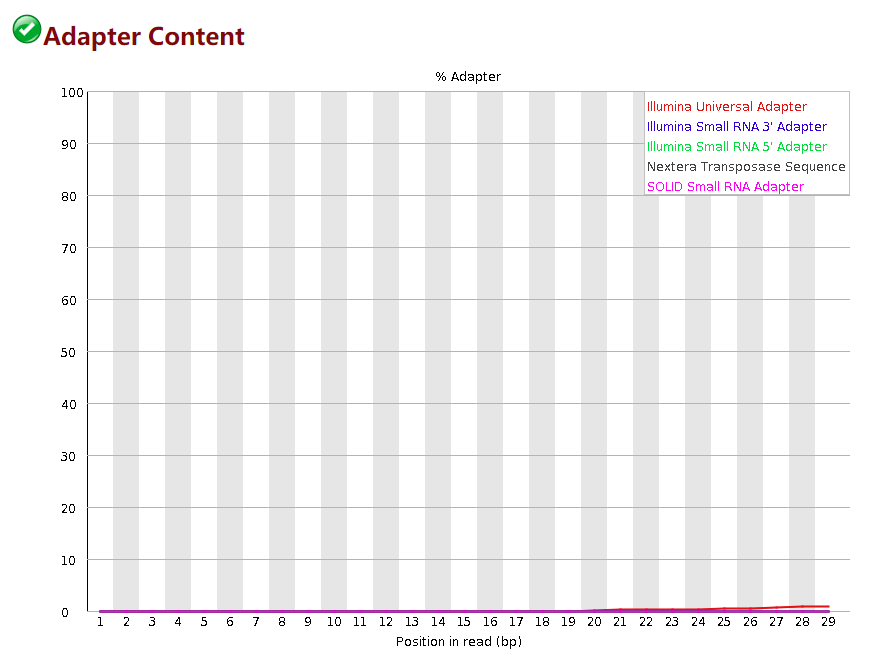
这个模块描述了接头的去除情况,横轴是碱基的位置,纵轴是是含有接头序列的比例。fastp默认是开启这个质控的不需要设置,如果你这个模块的质量很好可以用 -A 关掉质控,fastp是默认带有几个常用平台的接头序列。如果用默认的参数没有去除掉接头,你可以用 --adapter_fasta参数去指定你自己的接头序列,这个接头序列根据你测序平台不同会出现不同的接头。
到此为止,除了去除宿主污染其他所有的质控都做完了,宿主污染的质控方式留到下一章讲。
fastp的具体参数
usage: fastp -i <in1> -o <out1> [-I <in1> -O <out2>] [options...]
options:
# I/O options 即输入输出文件设置
-i, --in1 read1 input file name (string)
-o, --out1 read1 output file name (string [=])
-I, --in2 read2 input file name (string [=])
-O, --out2 read2 output file name (string [=])
-6, --phred64 indicates the input is using phred64 scoring (it'll be converted to phred33, so the output will still be phred33)
-z, --compression compression level for gzip output (1 ~ 9). 1 is fastest, 9 is smallest, default is 2. (int [=2])
--reads_to_process specify how many reads/pairs to be processed. Default 0 means process all reads. (int [=0])
# adapter trimming options 过滤序列接头参数设置
-A, --disable_adapter_trimming adapter trimming is enabled by default. If this option is specified, adapter trimming is disabled
-a, --adapter_sequence the adapter for read1. For SE data, if not specified, the adapter will be auto-detected. For PE data, this is used if R1/R2 are found not overlapped. (string [=auto])
--adapter_sequence_r2 the adapter for read2 (PE data only). This is used if R1/R2 are found not overlapped. If not specified, it will be the same as <adapter_sequence> (string [=])
# global trimming options 剪除序列起始和末端的低质量碱基数量参数
-f, --trim_front1 trimming how many bases in front for read1, default is 0 (int [=0])
-t, --trim_tail1 trimming how many bases in tail for read1, default is 0 (int [=0])
-F, --trim_front2 trimming how many bases in front for read2. If it's not specified, it will follow read1's settings (int [=0])
-T, --trim_tail2 trimming how many bases in tail for read2. If it's not specified, it will follow read1's settings (int [=0])
# polyG tail trimming, useful for NextSeq/NovaSeq data polyG剪裁
-g, --trim_poly_g force polyG tail trimming, by default trimming is automatically enabled for Illumina NextSeq/NovaSeq data
--poly_g_min_len the minimum length to detect polyG in the read tail. 10 by default. (int [=10])
-G, --disable_trim_poly_g disable polyG tail trimming, by default trimming is automatically enabled for Illumina NextSeq/NovaSeq data
# polyX tail trimming
-x, --trim_poly_x enable polyX trimming in 3' ends.
--poly_x_min_len the minimum length to detect polyX in the read tail. 10 by default. (int [=10])
# per read cutting by quality options 划窗裁剪
-5, --cut_by_quality5 enable per read cutting by quality in front (5'), default is disabled (WARNING: this will interfere deduplication for both PE/SE data)
-3, --cut_by_quality3 enable per read cutting by quality in tail (3'), default is disabled (WARNING: this will interfere deduplication for SE data)
-W, --cut_window_size the size of the sliding window for sliding window trimming, default is 4 (int [=4])
-M, --cut_mean_quality the bases in the sliding window with mean quality below cutting_quality will be cut, default is Q20 (int [=20])
# quality filtering options 根据碱基质量来过滤序列
-Q, --disable_quality_filtering quality filtering is enabled by default. If this option is specified, quality filtering is disabled
-q, --qualified_quality_phred the quality value that a base is qualified. Default 15 means phred quality >=Q15 is qualified. (int [=15])
-u, --unqualified_percent_limit how many percents of bases are allowed to be unqualified (0~100). Default 40 means 40% (int [=40])
-n, --n_base_limit if one read's number of N base is >n_base_limit, then this read/pair is discarded. Default is 5 (int [=5])
# length filtering options 根据序列长度来过滤序列
-L, --disable_length_filtering length filtering is enabled by default. If this option is specified, length filtering is disabled
-l, --length_required reads shorter than length_required will be discarded, default is 15. (int [=15])
# low complexity filtering
-y, --low_complexity_filter enable low complexity filter. The complexity is defined as the percentage of base that is different from its next base (base[i] != base[i+1]).
-Y, --complexity_threshold the threshold for low complexity filter (0~100). Default is 30, which means 30% complexity is required. (int [=30])
# filter reads with unwanted indexes (to remove possible contamination)
--filter_by_index1 specify a file contains a list of barcodes of index1 to be filtered out, one barcode per line (string [=])
--filter_by_index2 specify a file contains a list of barcodes of index2 to be filtered out, one barcode per line (string [=])
--filter_by_index_threshold the allowed difference of index barcode for index filtering, default 0 means completely identical. (int [=0])
# base correction by overlap analysis options 通过overlap来校正碱基
-c, --correction enable base correction in overlapped regions (only for PE data), default is disabled
# UMI processing
-U, --umi enable unique molecular identifer (UMI) preprocessing
--umi_loc specify the location of UMI, can be (index1/index2/read1/read2/per_index/per_read, default is none (string [=])
--umi_len if the UMI is in read1/read2, its length should be provided (int [=0])
--umi_prefix if specified, an underline will be used to connect prefix and UMI (i.e. prefix=UMI, UMI=AATTCG, final=UMI_AATTCG). No prefix by default (string [=])
--umi_skip if the UMI is in read1/read2, fastp can skip several bases following UMI, default is 0 (int [=0])
# overrepresented sequence analysis
-p, --overrepresentation_analysis enable overrepresented sequence analysis.
-P, --overrepresentation_sampling One in (--overrepresentation_sampling) reads will be computed for overrepresentation analysis (1~10000), smaller is slower, default is 20. (int [=20])
# reporting options
-j, --json the json format report file name (string [=fastp.json])
-h, --html the html format report file name (string [=fastp.html])
-R, --report_title should be quoted with ' or ", default is "fastp report" (string [=fastp report])
# threading options 设置线程数
-w, --thread worker thread number, default is 3 (int [=3])
# output splitting options
-s, --split split output by limiting total split file number with this option (2~999), a sequential number prefix will be added to output name ( 0001.out.fq, 0002.out.fq...), disabled by default (int [=0])
-S, --split_by_lines split output by limiting lines of each file with this option(>=1000), a sequential number prefix will be added to output name ( 0001.out.fq, 0002.out.fq...), disabled by default (long [=0])
-d, --split_prefix_digits the digits for the sequential number padding (1~10), default is 4, so the filename will be padded as 0001.xxx, 0 to disable padding (int [=4])
# duplication evaluation and deduplication
-D, --dedup enable deduplication to drop the duplicated reads/pairs
--dup_calc_accuracy accuracy level to calculate duplication (1~6), higher level uses more memory (1G, 2G, 4G, 8G, 16G, 24G). Default 1 for no-dedup mode, and 3 for dedup mode. (int [=0])
--dont_eval_duplication don't evaluate duplication rate to save time and use less memory.
# help
-?, --help print this message
参考链接
[2] fastp似乎无法处理per tile sequence quality

 浙公网安备 33010602011771号
浙公网安备 33010602011771号