
awk场景命令
# 注 mac 要用gwak代替awk.
~/Documents/materials/linux_shell
awk 'BEGIN{FS=OFS="\t"} NR==FNR{a[$3]=$1;b[$3]=$2} NR>FNR{$2=a[$1];$3=b[$1]; {print $2,$3,$1}' snp.raw snp.list >sub.txt #注意用BEGIN,各部分用{}括起来比较好。
awk 读取数据之前,运行BEGIN部分
所有数据读取后,推出awk前,运行END部分
注:
awk 一个文件遍历完成后,再遍历下一个文件
分号;表达式。
逗号, 并列 不换行
awk 处理每一行的字段内的数据,而默认的字段的分隔符为空格键或Tab键
$0表示某一行数据
$1第一列数据
$2第二列数据
...
Awk 模式-动作,如果动作省略,会默认输出符合模式(条件)的行,模式也可以省略。 正因为二者都可以省略,所以需要用大括号把动作括起来,以作区分。 模式决定动作什么时候执行。动作中多条语句用换行符或分号分开
Awk 动作中 print语句 由逗号,隔开的. 输出时默认用一个空格符号隔开
awk'条件类型{动作1}条件类型{动作2}...' filename
awk所有动作 单引号括住 ##其中{}括起多个命令
awk内在变量
NF:每一行所拥有的字段总数
$NF 即最后一列
NR:目前在第几行,从1开始,直到所有文件处理完 .注意不是$NR
FS:目前的分割字符,默认是空格键
NF 输入行的字段数量。 $NF:这行最后一列 ,$(NF-1)当前输入行的倒数第二个字段。
NR 到目前为止,读到的行的数量计数,从1开始,直到所有文件处理完
FNR 当前行在本文件的行数.注意不是$FNR
FS 输入行的字段分割符
OFS 输出字段分割符,默认是“ ”; ORS 输出记录的分割符,默认是“\n”
ARGC 命令行参数的个数
ARGV 命令行参数数组
ARGIND 当前被处理文件的ARGV标识符
可以给每个字段变量重新赋值,再调用如:
'BEGIN {FS=OFS=“\t”}. #BEGIN 动作设置FS与OFS
$4 == "North America" { $4 = "NA" }
$4 == "South America" { $4 = "SA" }
{ print } '
注:awk通常将字段数上限设置为100
print 用于简单快速的输出
Printf 格式化输出,尤其是print 拼接特殊字符如$1/stat/。如:{ printf"total pay for %s is $%.2f\n", $1, $2 * $3}
#awk -F '\t' 'print $0,$2\/stat\/' IgA.14390.dir.list 报错backslash not last character on line
awk -F '\t' '{printf "%s\t%s/stat/\n",$0,$2}' IgA.14390.dir.lis #运行顺利,留意末尾的\n
正则表达式 //匹配
正则表达式 [之后(里面)的^ 表示非 []是匹配只一个,1个以上得[]+,任意多即[]*,*表匹配0个或多个。
正则表示式[]里面的-表示范围 ,范围模式由逗号隔开的模式组成,如:pat1,pat2范围模式匹配多个输入行,从匹配pat1的行开始,到匹配pat2的行结束;如果part2一直没有匹配到,那么会自pat1匹配行开始一直输出到文件输入结束。
运算符: && ,||,!分别表示 且,或,非
内建变量FILENAME表示当前输入文件名
内建变量FNR表示从当前输入文件中,到目前为止读取到的行数。
如:awk 'FNR == 1,FNR == 5 { print FILENAME ":" $0 }' summary.py 输出1-5行 或 FNR<=5 {print $0}
awk 'FNR == 1||FNR == 5 { print FILENAME ":" $0 }' summary.py 输出第1行、5行
显示文件第1-3行 awk'NR==1,NR==3{print}' test.fa
FS 输入字段的分隔符,相当于-F默认为:space:
举例 awk -F ':'{print} test.fa 等同awk''BEGIN{FS=":"} test.fa
注:
- awk不需要声明变量类型,awk会根据上下文环境,推断出变量类型,如是字符串还是数字 如:100*$2 这里如果原$2不是数值,就会转换为数值。
- 如果字段变量引用到不存在的字段如总共4列,$5默认是空字符串或0。
匹配运算符~和!~ 如:awk '$0!~ /D2003005089/ {print $0}' optitye_45.sh >optitye_45_new.sh。 把不含样本D2003005089的行输入到optitye_45_new.sh
支持三元运算符,expr1 ? expr2 : expr3 如:{ print ($1 != 0 ? 1/$1 : "$1 is zero, line " NR) }
awk -F " " ' $NF>=5 {print $0}' light_cat_146.sh 把文件不存在的命令删除掉,不存在$5
注: awk 可以指定多个分隔符如awk -F '[,.]' '{print $(NF-3)".bam"}' cram_log.csv 就可以在保留路径情况下改后缀

awk 处理列后可以搭配paste -d ' ' cram.sh cram_log_target>cram_bam.sh 来按列合并两个文件
Awk的拼接符,即陆续写出字符串常量,变量,树组元素,函数返回值等。如{print NR “:” $0} #自定义变量和NR等内建变量不需要$
Awk 的正则表达式字符串,会对表达式的字符串自行求值后解释,不需像python一样先编译成正则表达式对象。如BEGIN { digits = "^[0-9]+$" }
$2 ~ digits
被双引号包围的字符串被awk解析时,起保护作用的反斜杠会被移除,所以还需要额外的反斜杠来保护它自己。如 $0 ~ /(\+|-)[0-9]+/ 与$0 ~ "(\\+|-)[0-9]+" 等价
awk 'BEGIN{print ARGV[1]}NR==1,NR==12 {print $1}' D2002002822.deduped.bam.type >>king7.type #打印文件名 ,然后打印1到12行内容
awk 比较两个值,如果都是数值,那么比较按数值进行。否则数值类型的操作数被强制转换成字符串,再按字符串的方式进行比较。
awk sub 是只替换一次,sub(r,s,t) 默认t是整个字符串,(如果指定t域如$1则就只在t指定域内)替换一次。相当于 sed 's//' 。如 awk '{ sub(/test/, "mytest", $1); print }' testfile
awk gsub 全局替换 ,gsub (regular expression, substitution string, target string) 默认是整个记录,如有指定第三个t域,则将替换t域内的r为s,相当于 sed 's//g' 。如:$ awk '{ gsub(/test/, "mytest", $1); print }' testfile
注:sub和gsub返回值都是替换次数,
如果要print出来替换次数就将替换表达式赋值给字段,
如果想print 替换结果不需单独赋值,
如:
echo "a b c 2011-11-22 a:d" | awk '$4=gsub(/-/,"",$4)' 返回 a b c 2 a:d
echo "a b c 2011-11-22 a:d" | awk 'gsub(/-/,"",$4) 返回 a b c 20111122 a:d
echo "a b c 2011-11-22 a:d" | awk '$5=gsub(/-/,"",$4)' 返回 a b c 20111122 2
substr(s,p,n) 返回s中从p位置开始长度为n的子字符串 如{ $1 = substr($1, 1, 3); print $0 } 注:没有指定n则将从p位置后所有字符串输出。
返回从起始位置起,指定长度之子字符串;若未指定长度,则返回从起始位置到字符串末尾的子字符串。
格式:
substr(s,p) 返回字符串s中从p开始的字符串部分
substr(s,p,n) 返回字符串s中从p开始长度为n的字符串部分
例子:
echo "123" | awk '{print substr($0,1,1)}
zcat input_file.fastq.gz | awk 'NR%4==1{printf ">%s\n", substr($0,2)}NR%4==2{print}' > output_file.fa #以字符串形式打印输出变量str后光标换行
awk通过花括号用于语句组合,实现流程控制。如:
if (expression) statements1
else
statements2
例子:
lastlog |awk '{if ($4=="in**") print $1,"\t",$2,$3,$4; else print $1"\t"$9"-"$5"-"$6,$7}' >lastlog.txt
常用的计算方式:
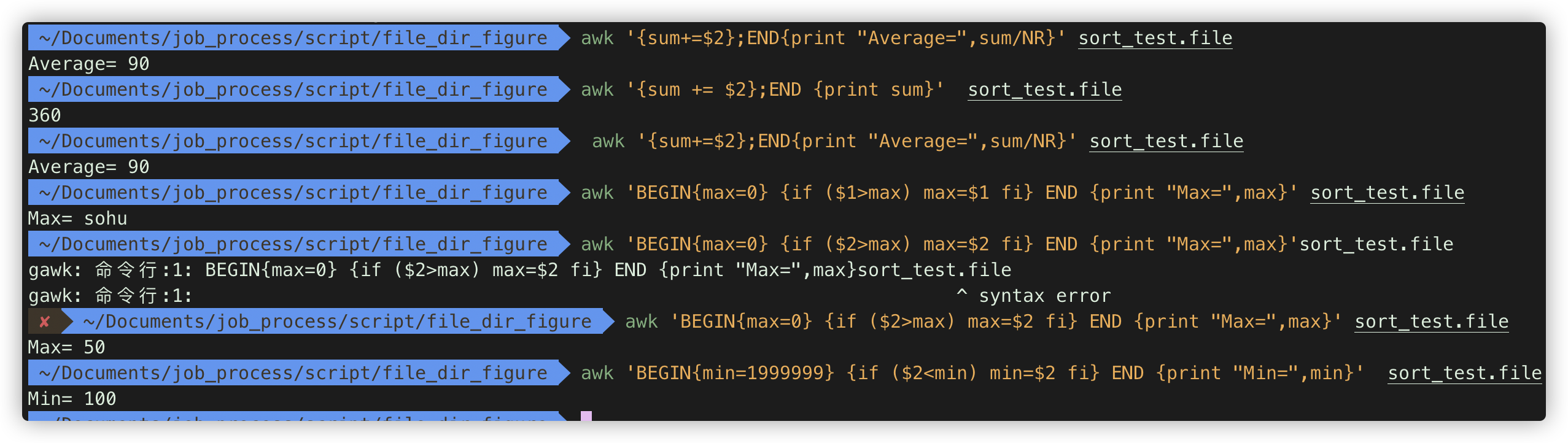
计算第二列加和。 awk '{sum += $2};END {print sum}' sort_test.file
计算平均值 awk '{sum+=$2};END{print "Average=",sum/NR}' sort_test.file
最大值 awk 'BEGIN{max=0} {if ($2>max) max=$2 fi} END {print "Max=",max}' sort_test.file
最小值 awk 'BEGIN{min=1999999} {if ($2<min) min=$2 fi} END {print "Min=",min}' sort_test.file
awk '{if (NR%4==2|NR%4==0){print substr($0,1,50)}else{print $0}}'your.fq #取fastq文件5‘端前50个base的命令;
awk '{if (NR%4==2||NR%4==0){print substr(($0),length($0)-50+1)}}'your.fq #取3’端后50个base
zcat CL100006359_L01_1_1.fq.gz |awk '{if (NR%4==2){print length($0)}}' #数fq.gz read碱基数 :
cat 20171231_SZ_hfpc_info.csv | awk -F "," '{print $1}' |uniq -c -d #判断某域是否有重复值
注: uniq -c 在行前方显示重复次数 ;-d 仅显示重复的行
Awk 可以作为管道符号接在其他命令后面,输出一部分内容。
bamsex 例子
result=$(awk -v OFS="\t" 'NR<=22{genome+=$2;sum+=$3}(NR==23||NR==24){cov[$1]=100*$3/$2}END{autocov=100*sum/genome;print autocov,cov["chrX"],cov["chrY"],cov["chrX"]/autocov,cov["chrY"]/autocov}' $idxstats)
# awk 默认变量的初始值是0,不需要初始化变量。
可进一步参考 http://www.zsythink.net/archives/tag/awk/
awk 数组下标可是字符也可是数字,使用前数组可以不用先定义,可以直接执行++(计数),+=(字符串) 如:
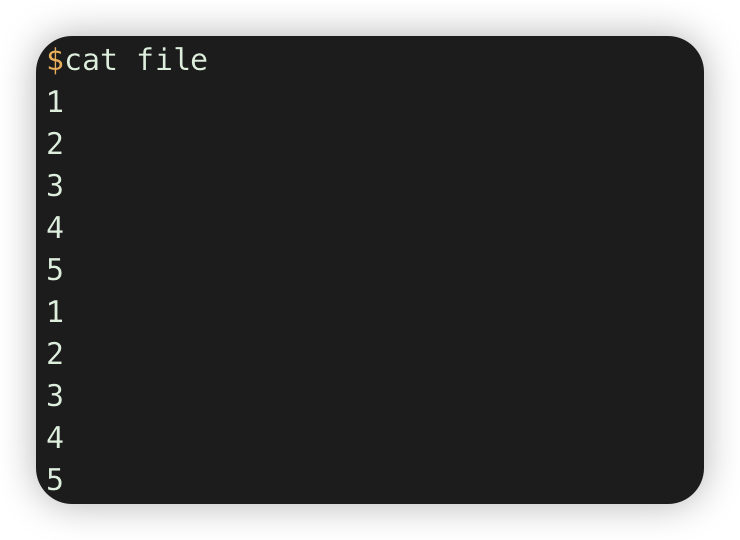
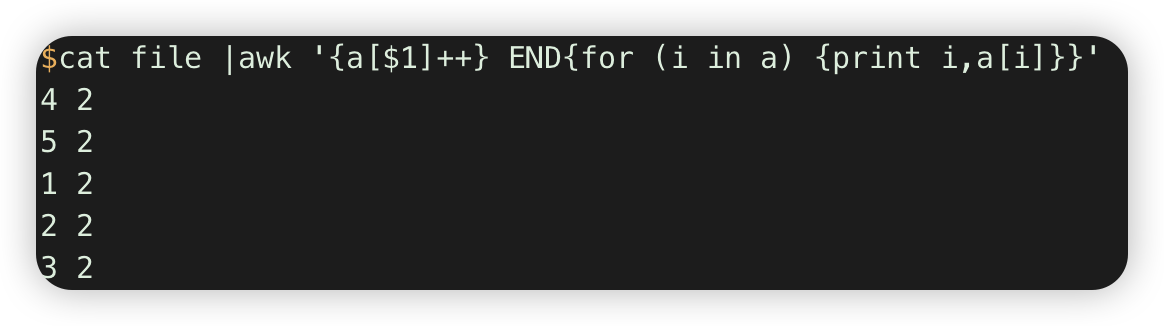
seq 5 >file
sed 5 >>file
cat file |awk '{a[$1]++} END{for (i in a) {print i,a[i]}}'. # 计数每行重复的次数
搭配 NR, FNR 使用
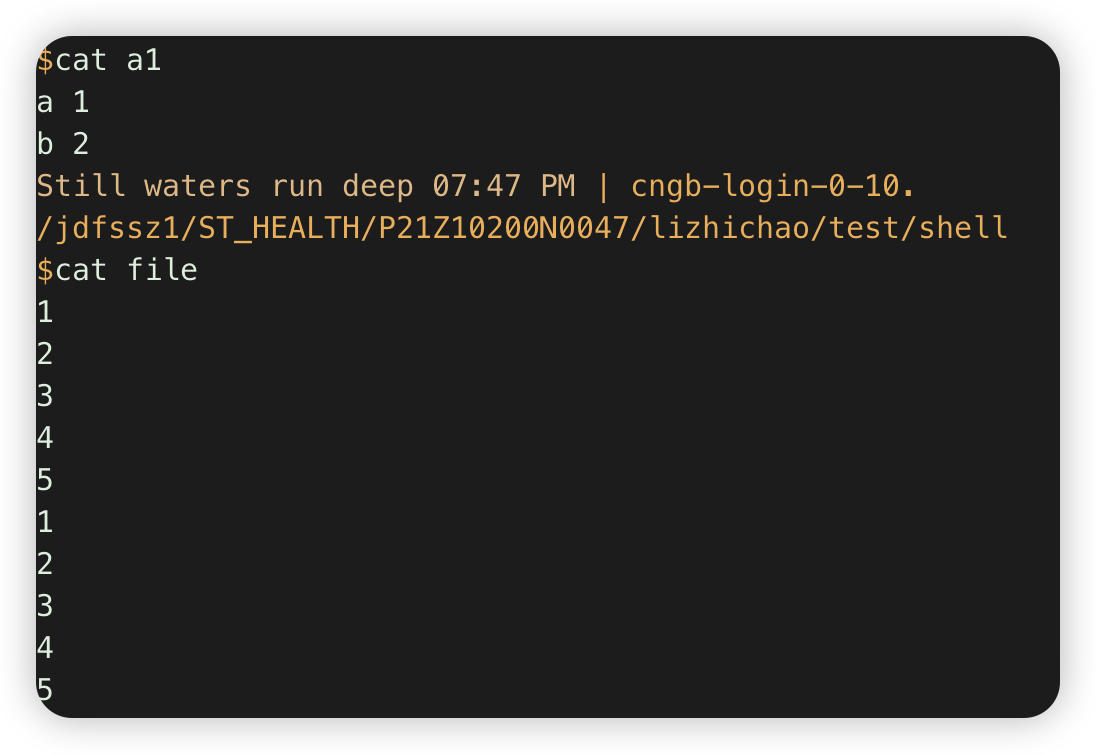
1.合并多个多文件,第一个文件带header,其他的不带
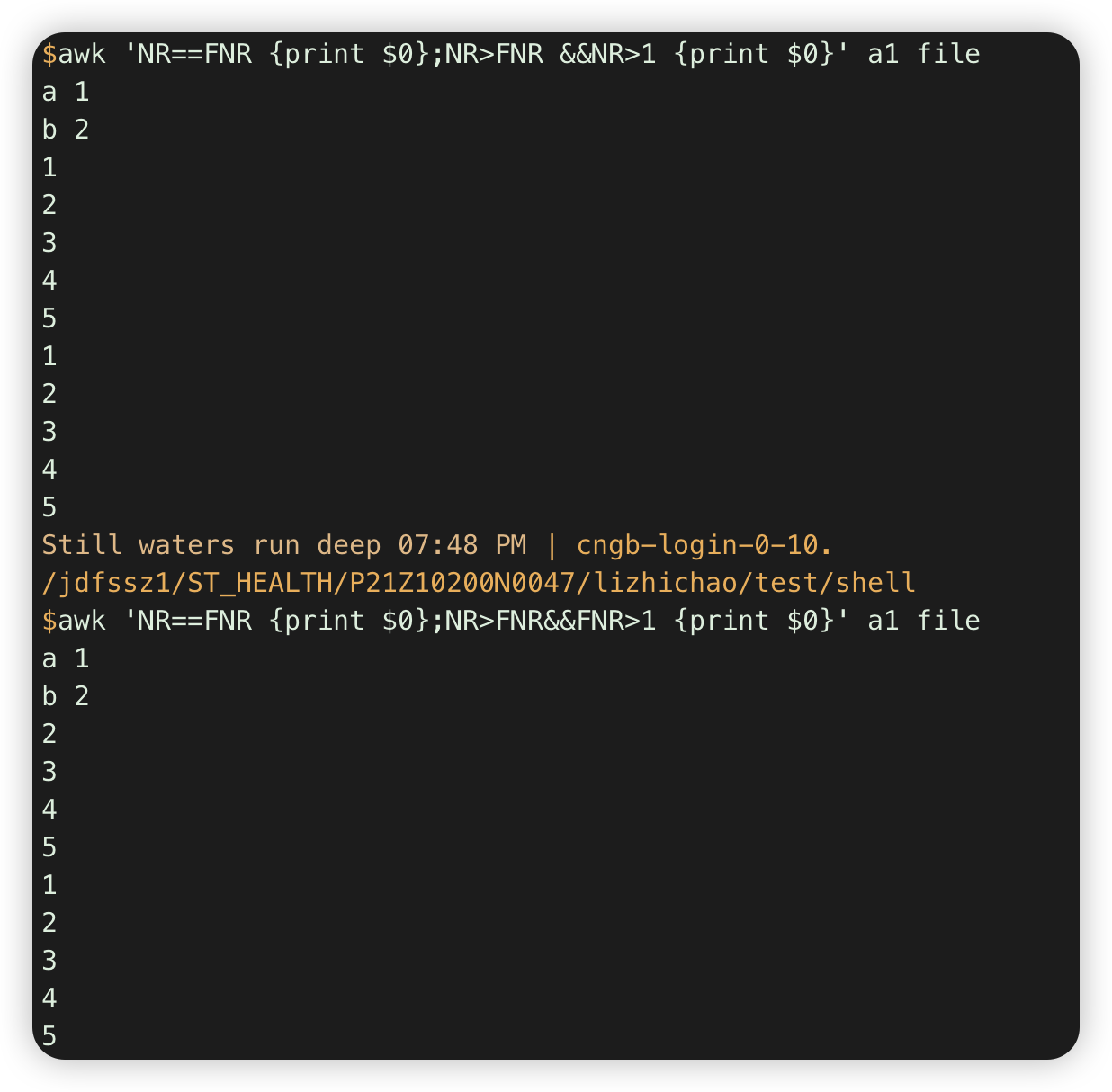
awk 'NR==FNR {print $0};NR>FNR&&FNR>1 {print $0}' a1 file #al全部行,file行跳过第一行
注意:通过FNR判断在当前文件第几行
等于是==, 不是=
结合循环、字符串拼接生成要合并的文件列表
a=""
$while read line
> do
> a="$a conditional_$line.jma.cojo"
> done<IgA13756.sex_age.PHENO1.glm.logistic.1e-6_chr.csv.uniq
awk 'BEGIN{FS=OFS="\t"} NR=FNR{print $0};NR>FNR&&FNR>1{print $0}' $a
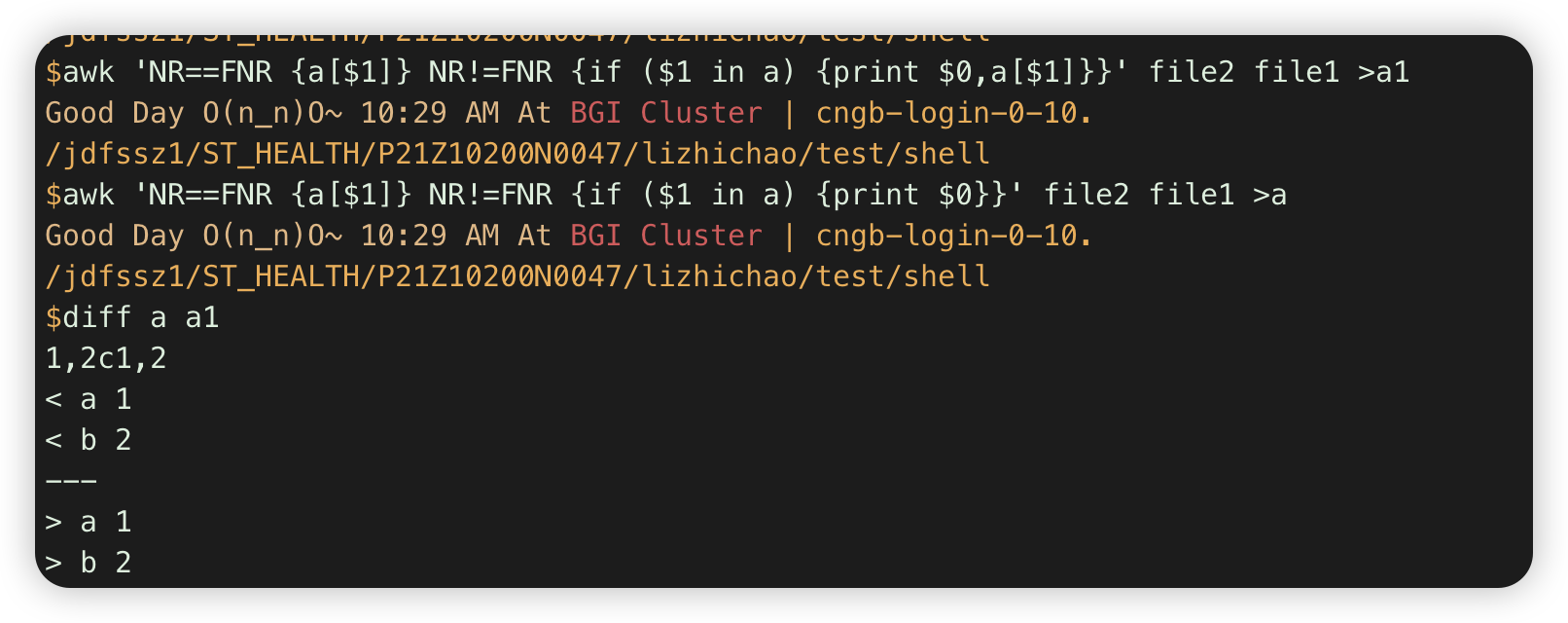
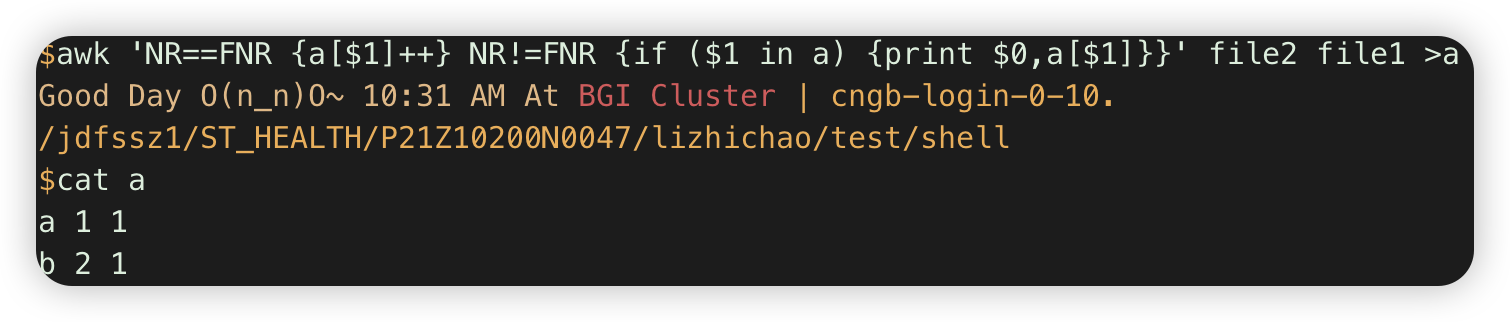
2.awk 'NR==FNR {a[$1]} NR!=FNR {if ($1 in a) {print $0}}' file2 file1 #第一个文件存在字典,第二个文件输出与之overlap的行 .
注:这里a[$1]没++,所以默认不是值,是空字符串blank

awk 搭配next ,是用于跳过本行的后续表达式计算,跳到下一行开始,节省时间
如 awk ‘$4<=20 {printf "%s\t%s\n",$0,"*";next} $4>20 {print $0;}’ food_list.txt
awk 'BEGIN{FS=OFS="\t"}{for(i=3;i<NF;i++)printf("%s\t",$i);print $NF}' IgA13474.covar.pc_sex.snp_only.PHENO1.glm.logistic_RS >IgA13474.covar.pc_sex.snp_only.PHENO1.glm.logistic_RS.abondon_chr_pos. #删除前两列,输出tab分隔的文件
本文来自博客园,作者:BioinformaticsMaster,转载请注明原文链接:https://www.cnblogs.com/koujiaodahan/p/15754299.html

posted on 2021-12-31 19:57 BioinformaticsMaster 阅读(294) 评论(0) 编辑 收藏 举报


【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· 基于Microsoft.Extensions.AI核心库实现RAG应用
· Linux系列:如何用heaptrack跟踪.NET程序的非托管内存泄露
· 开发者必知的日志记录最佳实践
· SQL Server 2025 AI相关能力初探
· Linux系列:如何用 C#调用 C方法造成内存泄露
· 震惊!C++程序真的从main开始吗?99%的程序员都答错了
· 【硬核科普】Trae如何「偷看」你的代码?零基础破解AI编程运行原理
· 单元测试从入门到精通
· 上周热点回顾(3.3-3.9)
· winform 绘制太阳,地球,月球 运作规律