
Drop-seq测序平台dge.txt.gz格式转化成h5格式
dge.txt.gz格式简介
dge.txt.gz格式是Drop-seq format(一个单细胞RNA测序平台,三种常见基于液滴的单细胞RNA测序平台10X Genomics Chromium、inDrop和Drop-seq),也可能命名为.digital_expression.txt.gz。
Drop-seq测序平台
dge.txt格式转化成h5格式
因为这个格式确实少见,所以把遇到可能的方案都记录下来了,以供启示。
我的方法
安装
- conda安装Seurat:
conda install -c bioconda r-seurat# 如果不成功,这个包挺好安装的,自己解决吧 - conda安装 SeuratDisk:
conda install -c pwwang r-seuratdisk# 这个包不易安装,建议使用该命令,有冲突更改其他包
使用
## Read File
Patient_scRNA.counts <- read.delim("/home/xxx/downloads/XXXXXX_S1_dge.txt", row.names = 1)
## Create Seurat Object
library(Seurat)
library(SeuratDisk)
Patient_OC <- CreateSeuratObject(counts = Patient_scRNA.counts, project = "XXXXX")
## Save File
SaveH5Seurat(Patient_OC, filename = "/home/xxxxx/downloads/h5name.h5Seurat")
Convert("/home/xxxxx/downloads/h5name.h5Seurat", dest = "h5ad")
sceasy 包
sceasy 包 提供了seurat、anndata、loom、SingleCellExperiment四种格式间的转换,但是我没搞明白怎么用,如果有生物基础的可以尝试。
安装sceasy
建议创建新的conda环境,R语言版本为4.0~4.1,安装有问题可以留言,我安装成功了
- 安装sceasy
sceasy 可以作为 bioconda 包安装:conda install -c bioconda r-sceasy
或者作为 R 包:devtools::install_github("cellgeni/sceasy")
这将需要双导体包 BiocManager 和 LoomExperiment:
注意: BiocManager需要与R语言版本对应
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(c("LoomExperiment", "SingleCellExperiment"))
-
安装 anndata 包:
conda install anndata -c bioconda -
安装 reticulate 包:
install.packages('reticulate')
如果计划在 loom 和 anndata 之间进行转换,请确保loompy已安装该包:conda install loompy -c bioconda
使用
下面的使用没有搞明白怎么用,一直报错unable to find an inherited method for function ‘assayNames’ for signature ‘"Seurat"’
seurat、anndata、loom、SingleCellExperiment四种格式间的转换:
注意: 不同对象转换时的输入是文件还是 object
# 使用这个,(但是不清楚sce_object数据如何读取)
sceasy::convertFormat(sce_object, from="sce", to="anndata",
outFile='filename.h5ad')
# 其他格式的转化:
sceasy::convertFormat(seurat_object, from="seurat", to="anndata",
outFile='filename.h5ad')
sceasy::convertFormat(h5ad_file, from="anndata", to="seurat",
outFile='filename.rds')
sceasy::convertFormat(seurat_object, from="seurat", to="sce",
outFile='filename.rds')
sceasy::convertFormat(sce_object, from="sce", to="loom",
outFile='filename.loom')
sceasy::convertFormat('filename.loom', from="loom", to="anndata",
outFile='filename.h5ad')
sceasy::convertFormat('filename.loom', from="loom", to="sce",
outFile='filename.rds')
scCustomize(似乎可行)
因为这个格式确实少见,所以把遇到可能的方案都记录下来了,以供启示。
官方介绍
导入带文件前缀的分隔矩阵单目录
数据通常会以包含所有信息的单个文件(.csv、.tsv、.txt 等)的形式上传到 NCBI GEO 或其他存储库。
在此示例中,我将使用 Hammond 等人于 2019 年 ( Immunity ) 提供的数据,这些数据是从NCBI GEO GSE121654下载的。
Read_GEO_Delim使用 fread 函数自动检测文件分隔符并快速读取,然后将对象转换为稀疏矩阵以节省内存
# Read in and use file names to name the list (default)
GEO_Single <- Read_GEO_Delim(data_dir = "assets/GSE121654_RAW_Hammond/GSE121654_RAW_Hammond/", file_suffix = ".dge.txt.gz")
# Read in and use new sample names to name the list
GEO_Single <- Read_GEO_Delim(data_dir = "assets/GSE121654_RAW_Hammond/GSE121654_RAW_Hammond/", file_suffix = ".dge.txt.gz",
sample_names = c("sample01", "sample02", "sample03", "sample04"))
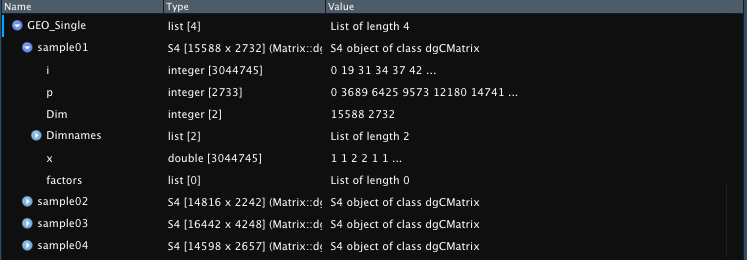
根据文件名或提供的 sample_names参数以默认命名的示例输出。
Read_GEO_Delim附加参数,请参阅手动输入以了解更多信息。
