图解微生物组数据分析QIIME 2平台笔记(三)
本文内容参考自:QIIME2中文帮助文档 (Chinese Manual);QIIME 2 user documentation — QIIME 2 2024.5.0 documentation
因QIIME2 的持续更新原文章内容的代码部分不可用,现写新版QIIME2内容源码笔记如下
本文内容源码均是在Oracle VM VirtualBox 虚拟机创建的QIIME平台中于2024年9月16日测试修改完成
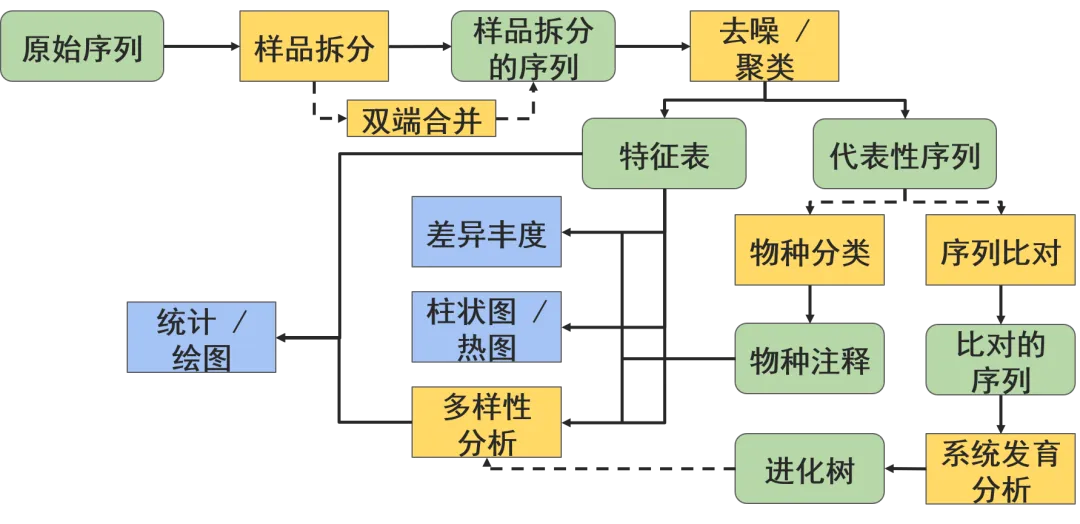
一、16S RNA测序数据上游分析
(1)前期准备
#获取源数据
# 创建并进入工作目录
mkdir -p qiime2-moving-pictures-tutorial
cd qiime2-moving-pictures-tutorial
# 下载实验设计数据(需要FQ下载,也可关注公众号"随笔茶屋",回复QIIME2 获取所需源数据)
wget -O "sample-metadata.tsv" "https://data.qiime2.org/2017.7/tutorials/moving-pictures/sample_metadata.tsv"
# 下载实验测序数据
mkdir -p emp-single-end-sequences
wget -O "emp-single-end-sequences/barcodes.fastq.gz" "https://data.qiime2.org/2017.7/tutorials/moving-pictures/emp-single-end-sequences/barcodes.fastq.gz"
wget -O "emp-single-end-sequences/sequences.fastq.gz" "https://data.qiime2.org/2017.7/tutorials/moving-pictures/emp-single-end-sequences/sequences.fastq.gz"
# 生成qiime需要的artifact文件(qiime文件格式,将原始数据格式标准化)
qiime tools import \ #用于导入数据
--type EMPSingleEndSequences \ #指定数据类型,EMPSingleEndSequences表示单端序列数据,这是微生物生态学研究中常见的一种数据类型
--input-path emp-single-end-sequences \ #指定了原始数据文件的路径
--output-path emp-single-end-sequences.qza #指定了转换的artifact文件的存储路径
#样品拆分
# 使用QIIME 2的demux emp-single插件进行样品的去复用操作
# 这个步骤将根据样品的条形码将混合的序列数据拆分到对应的样品中
qiime demux emp-single \
# 指定输入的单端序列数据文件(.qza格式的QIIME 2 artifact)
--i-seqs emp-single-end-sequences.qza \
# 指定包含样品条形码信息的元数据文件
--m-barcodes-file sample-metadata.tsv \
# 指定元数据文件中包含条形码序列的列名
--m-barcodes-column BarcodeSequence \
# 指定输出文件的名称和路径,去复用后的序列数据将会保存在这个文件中
--o-per-sample-sequences demux-sequences.qza \
#指定了输出的条形码错误校正详情。
--o-error-correction-details error-correction-details.qza
# 结果统计
qiime demux summarize \
--i-data demux-sequences.qza \
--o-visualization demux-sequences.qzv
# 查看结果 (依赖XShell+XManager或其它ssh终端和图形界面软件)
qiime tools view demux-sequences.qzv
样品拆分的数据结果如下:
在线查看网站file:///tmp/qiime2-archive-hv2f3aen/d5ac6f44-80a0-4020-bccf-56b49222c199/data/index.html
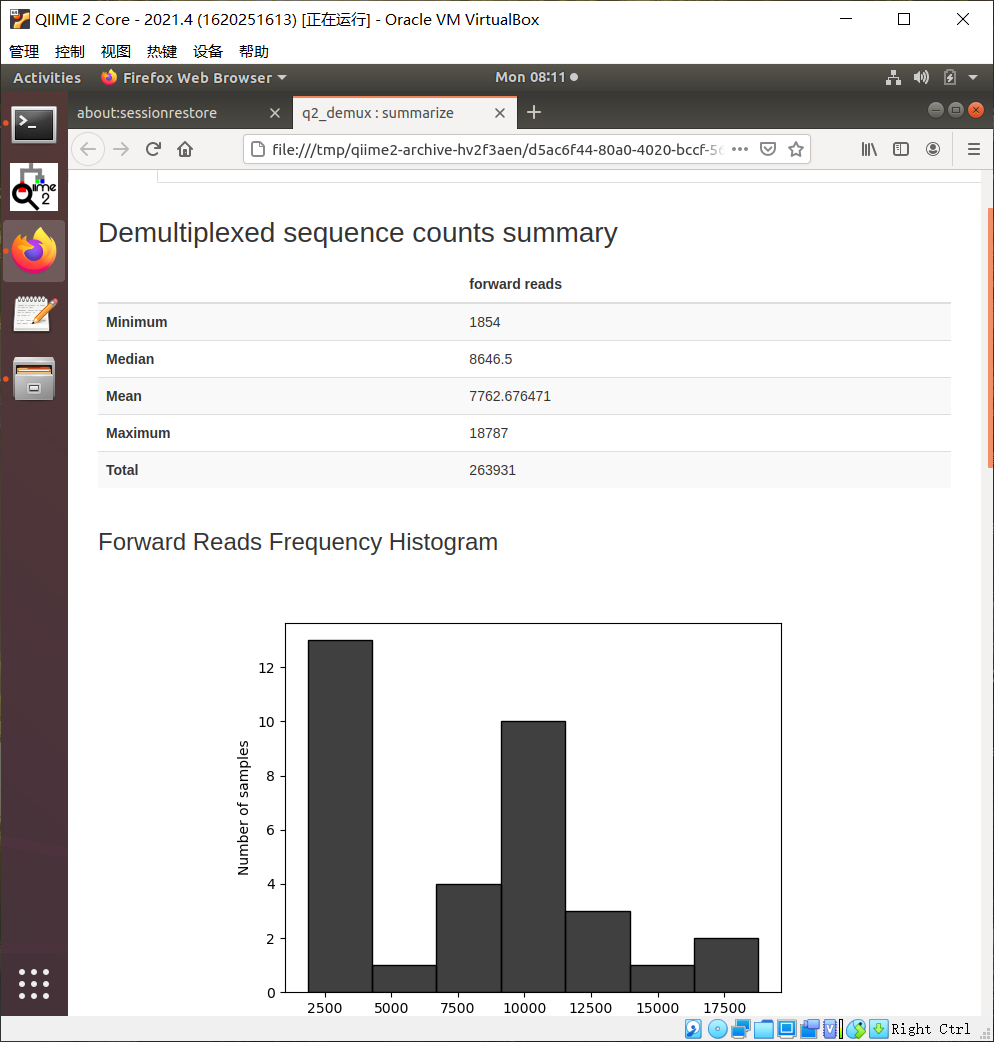
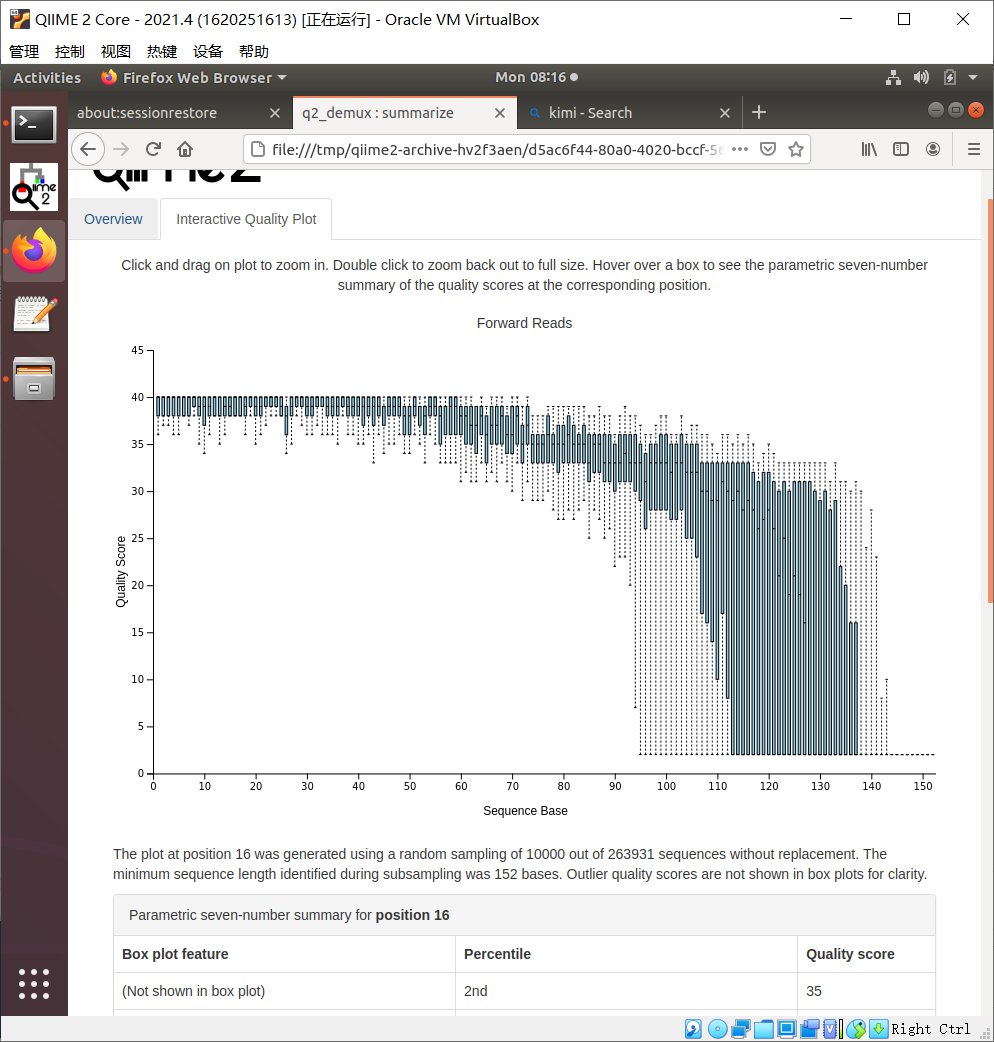
注:基于上图样品拆分的结果可以看出左端质量较高,无低质量区,右端从大于120之后质量下降极大;
附:qiime 中demux的部分选项如下所示(可忽略)
Usage: qiime demux emp-single [OPTIONS]
Demultiplex sequence data (i.e., map barcode reads to sample ids) for data
generated with the Earth Microbiome Project (EMP) amplicon sequencing
protocol. Details about this protocol can be found at
http://www.earthmicrobiome.org/protocols-and-standards/
Inputs:
--i-seqs ARTIFACT RawSequences | EMPSingleEndSequences |
EMPPairedEndSequences
The single-end sequences to be demultiplexed.
[required]
Parameters:
--m-barcodes-file METADATA
--m-barcodes-column COLUMN MetadataColumn[Categorical]
The sample metadata column containing the per-sample
barcodes. [required]
--p-golay-error-correction / --p-no-golay-error-correction
Perform 12nt Golay error correction on the barcode
reads. [default: True]
--p-rev-comp-barcodes / --p-no-rev-comp-barcodes
If provided, the barcode sequence reads will be
reverse complemented prior to demultiplexing.
[default: False]
--p-rev-comp-mapping-barcodes / --p-no-rev-comp-mapping-barcodes
If provided, the barcode sequences in the sample
metadata will be reverse complemented prior to
demultiplexing. [default: False]
--p-ignore-description-mismatch / --p-no-ignore-description-mismatch
If enabled, ignore mismatches in sequence record
description fields. [default: False]
Outputs:
--o-per-sample-sequences ARTIFACT SampleData[SequencesWithQuality]
The resulting demultiplexed sequences. [required]
--o-error-correction-details ARTIFACT ErrorCorrectionDetails
Detail about the barcode error corrections. [required]
Miscellaneous:
--output-dir PATH Output unspecified results to a directory
--verbose / --quiet Display verbose output to stdout and/or stderr during
execution of this action. Or silence output if
execution is successful (silence is golden).
--examples Show usage examples and exit.
--citations Show citations and exit.
--help Show this message and exit.
#序列质控和生成OTU表
可以选择选择使用DADA2或Deblur 两种方法,去除低质量的序列和生成OTU表,这里推荐使用DADA21.选择DADA2
# 单端序列去噪, 去除左端0bp(--p-trim-left用于切除边缘低质量区),序列切成120bp长;生成代表序列和OTU表;并重命名用于下游分析
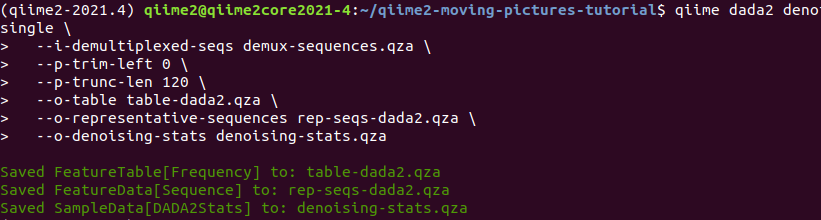
qiime dada2 denoise-single \
--i-demultiplexed-seqs demux-sequences.qza \
--p-trim-left 0 \ #--p-trim-left 截取左端低质量序列,我们看上图中箱线图,左端质量都很高,无低质量区,设置为0;
--p-trunc-len 120 \ #序列截取长度,也是为了去除右端低质量序列,我们看到大于120以后,质量下降极大,甚至中位数都下降至20以下,需要全部去除
--o-representative-sequences rep-seqs-dada2.qza \ #生成裁剪后的代表序列
--o-table table-dada2.qza #生成OTU特征表
--o-denoising-stats denoising-stats.qza #指定了输出的去噪统计信息
#使用mv命令对文件进行重命名
mv rep-seqs-dada2.qza rep-seqs.qza
mv table-dada2.qza table.qza
2.选择Deblur
# 按测序质量过滤序列
qiime quality-filter q-score \
--i-demux demux-sequences.qza \
--o-filtered-sequences demux-filtered.qza \
--o-filter-stats demux-filter-stats.qza
# 去冗余生成OTU表和代表序列;结果文件名有deblur,没有用于下游分析,请读者想测试的自己尝试
qiime deblur denoise-16S \
--i-demultiplexed-seqs demux-filtered.qza \
--p-trim-length 120 \
--o-representative-sequences rep-seqs-deblur.qza \
--o-table table-deblur.qza \
--o-stats deblur-stats.qza
截止目前qiime2-moving-pictures-tutorial文件夹包含的文件如下
#OTU Feature 表统计
qiime feature-table summarize \#生成摘要统计信息
--i-table table.qza \#输入OTU特征表文件
--o-visualization table.qzv \
--m-sample-metadata-file sample-metadata.tsv#指定样本元数据文件
qiime tools view table.qzv # 展打开网页版统计结果,看下图
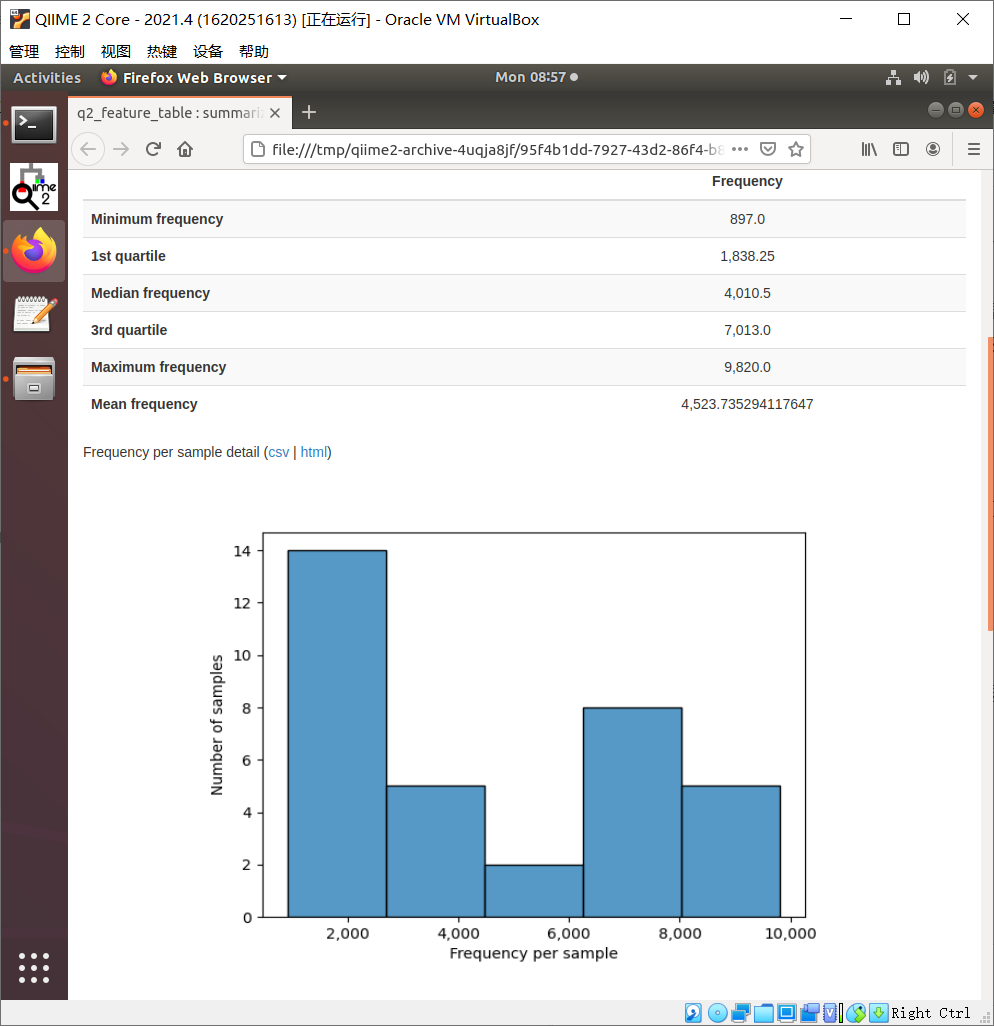
注:最小的序列值为897
#代表序列统计
qiime feature-table tabulate-seqs \
--i-data rep-seqs.qza \
--o-visualization rep-seqs.qzv
qiime tools view rep-seqs.qzv # 展打开网页版统计结果,看下图
(2)多样性分析
注:多样性分析包括基于系统发育树的多样性,常用命令qiime diversity core-metrics;专注于非系统发育多样性指标#建立用于多样性分析的有根树(基于系统发育树的多样性计算)
# 通过MAFFT算法对代表性序列(rep-seqs.qza)进行多序列比对。比对结果被保存到aligned-rep-seqs.qza文件中。
qiime alignment mafft \
--i-sequences rep-seqs.qza \
--o-alignment aligned-rep-seqs.qza
# 对已比对的序列(aligned-rep-seqs.qza)进行掩蔽,移除高变区
qiime alignment mask \
--i-alignment aligned-rep-seqs.qza \
--o-masked-alignment masked-aligned-rep-seqs.qza
# 基于掩蔽后的序列(masked-aligned-rep-seqs.qza)构建一棵无根树
qiime phylogeny fasttree \
--i-alignment masked-aligned-rep-seqs.qza \
--o-tree unrooted-tree.qza
# 无根树转换为有根树
qiime phylogeny midpoint-root \
--i-tree unrooted-tree.qza \
--o-rooted-tree rooted-tree.qza
Alpha多样性和Beta多样性分析
+ Alpha 多样性 - 香农多样性指数(群落丰富度的定量衡量) - 观测特征(群落丰富度的定性度量) - Faith's Phylogenetic Diversity(群落丰富度的定性衡量标准,包含特征之间的系统发育关系) - 均匀度(或 Pielou 的均匀度;社区均匀度的度量) + Beta 多样性 - Jaccard 距离(社区差异的定性度量) - Bray-Curtis 距离(群落相异性的定量度量) - 未加权 UniFrac 距离(一种群落不相似性的定性测量,包含特征之间的系统发育关系) - 加权 UniFrac 距离(一种群落不相似性的定量测量,包含特征之间的系统发育关系#-------------------------------------------------
# 计算Alpha多样性的特定指标
qiime diversity alpha \
--i-table table.qza \
--p-metric observed_features \#这里指定的是特征数(OTUs),还可以指定shannon、faith_pd等
--o-alpha-diversity alpha-diversity.qza
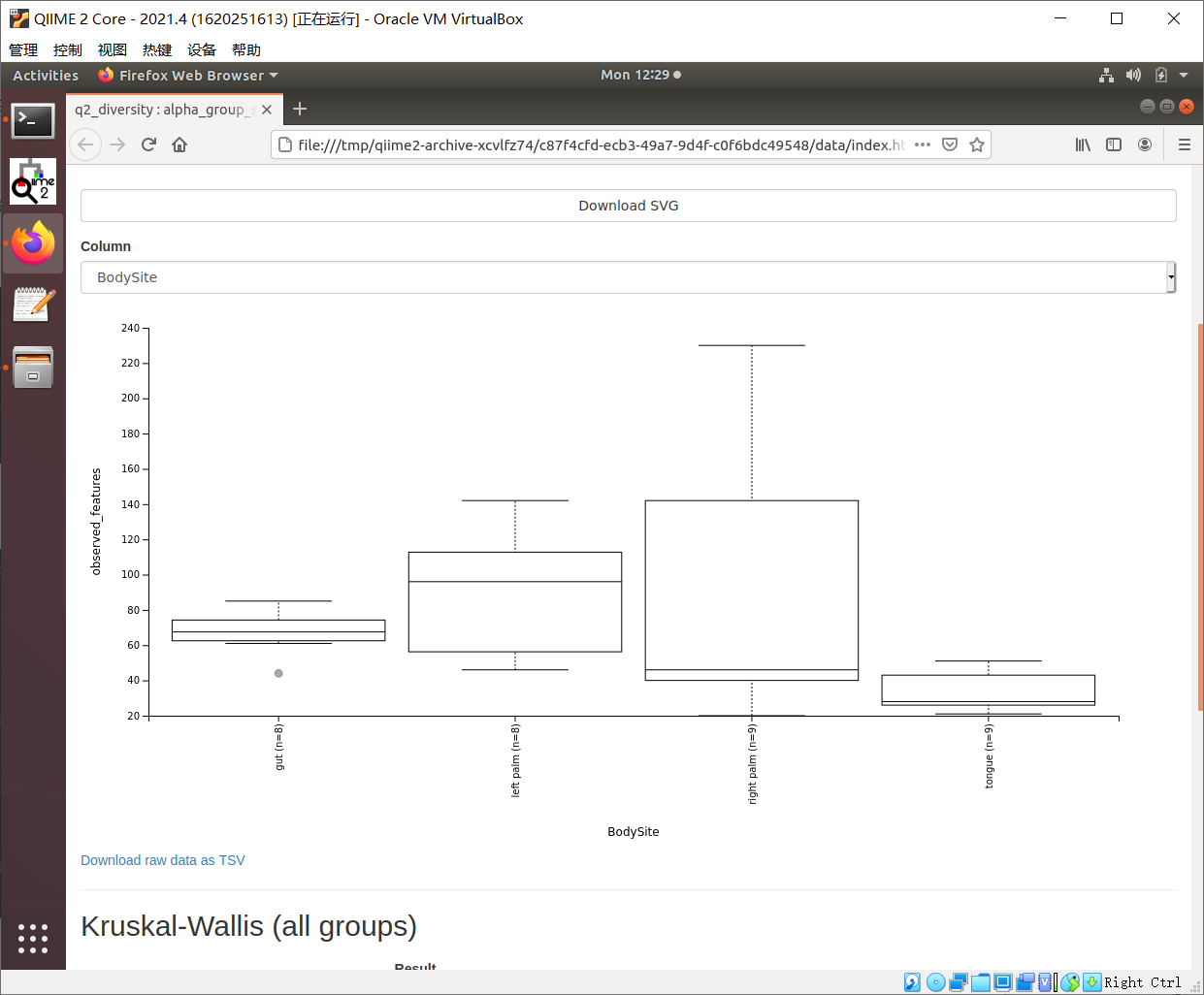
# 统计Alpha多样性组间差异是否显著
qiime diversity alpha-group-significance \
--i-alpha-diversity alpha-diversity.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization alpha-diversity-group-significance.qzv
# 网页展示结果,只要是qzv的文件,均可用qiime tools view查看或在线https://view.qiime2.org/查看,以后不再赘述
qiime tools view alpha-diversity-group-significance.qzv
#---------------------------------------------------
#基于系统发育树的命令创建faith_pd指数的可视化结果
qiime diversity alpha-phylogenetic \
--i-table table.qza \
--i-phylogeny rooted-tree.qza \
--p-metric faith_pd \
--o-alpha-diversity alpha-diversity.qza
#导出Faith_pd指数的结果
qiime tools export --input-path alpha-diversity.qza --output-path exported-alpha-diversity
#----------------------------------------------------
#多样性指标的计算
qiime diversity core-metrics-phylogenetic \
--i-phylogeny rooted-tree.qza \
--i-table table.qza \
--p-sampling-depth 1103 \
--m-metadata-file sample-metadata.tsv \
--output-dir core-metrics-results
#计算了多样性指标之后,我们可以开始在样本元数据的上下文中探索样本的微生物组成。
#测试分类元数据列与 alpha 多样性数据之间的关联。我们将在这里对 Faith Phylogenetic Diversity(衡量社区丰富度的指标)和均匀度指标进行此操作
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/faith_pd_vector.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization core-metrics-results/faith-pd-group-significance.qzv
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/evenness_vector.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization core-metrics-results/evenness-group-significance.qzv
# 使用 QIIME 2 的 diversity 插件中的 beta-group-significance 命令
# 该命令用于测试不同样本组之间的 beta 多样性是否具有统计学上的显著差异
qiime diversity beta-group-significance \
# --i-distance-matrix:指定输入的距离矩阵文件
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza \
# --m-metadata-file:指定样本元数据文件
--m-metadata-file sample-metadata.tsv \
# --m-metadata-column:指定用于分组的元数据列
--m-metadata-column BodySite \
# --o-visualization:指定输出的可视化文件路径
--o-visualization core-metrics-results/unweighted-unifrac-body-site-significance.qzv \
# --p-pairwise:指定是否进行成对比较
--p-pairwise
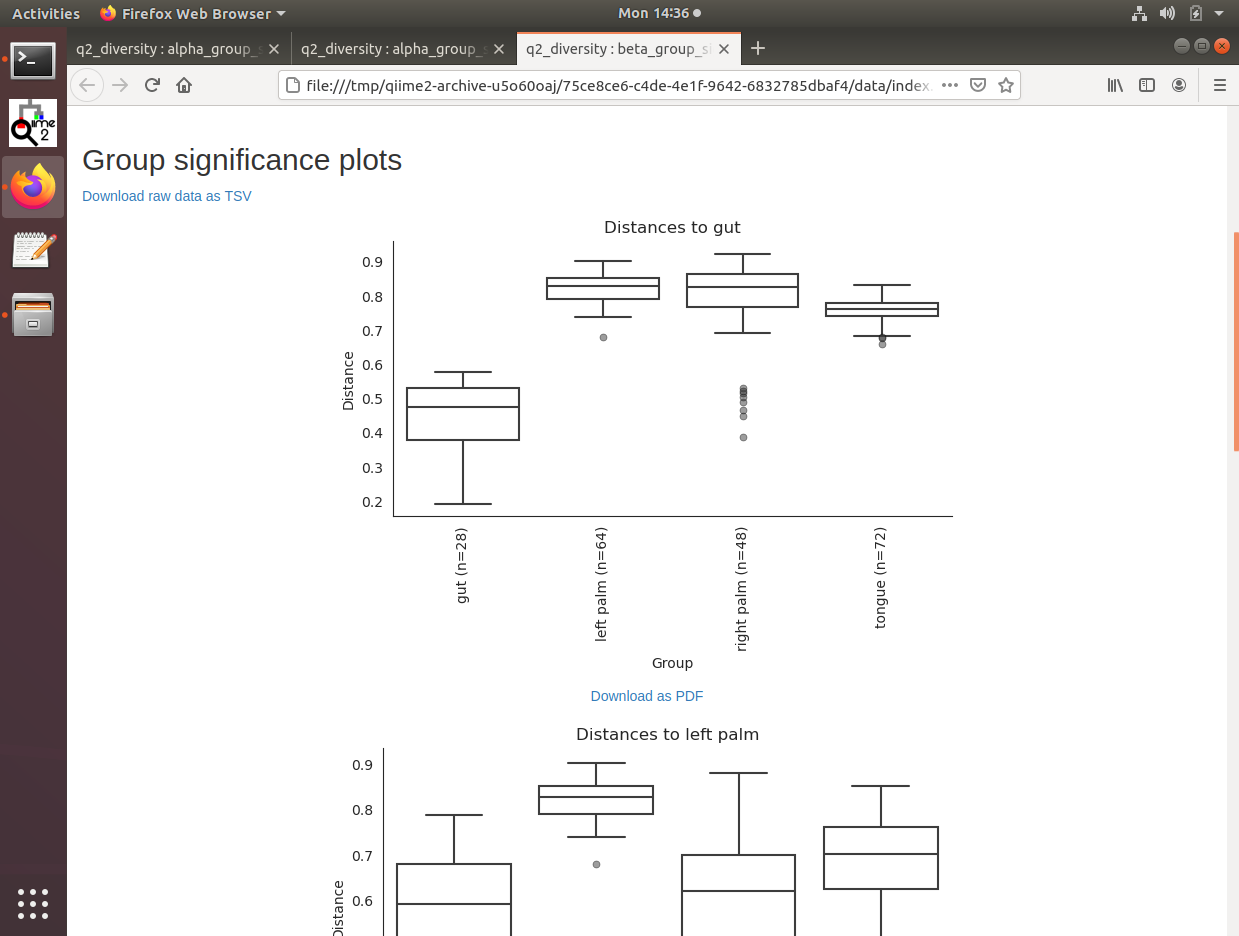
qiime emperor plot \
--i-pcoa core-metrics-results/unweighted_unifrac_pcoa_results.qza \
--m-metadata-file sample-metadata.tsv \
--p-custom-axes DaysSinceExperimentStart \
--o-visualization core-metrics-results/unweighted-unifrac-emperor-days-since-experiment-start.qzv

 浙公网安备 33010602011771号
浙公网安备 33010602011771号