

6.3 Using Accelerated Molecular Dynamics (aMD) to enhance sampling
英文官网:http://ambermd.org/tutorials/advanced/tutorial22/index.php
另外可参考 https://blog.chembiosim.com/aMD_in_Amber/
传统的分子动力学允许人们访问数十到数百纳秒的时间尺度; 然而,许多感兴趣的生物过程发生在长达几毫秒或更长时间的较长时间尺度上。
加速动力学是对势能的一种修改——在模拟时减少局域barriers的高度,允许计算得更快。aMD是一个有趣的选择,因为它只需要单个副本的进化,并且它不需要任何关于势能面的先前知识。
aMD 有两种主要版本,一种提高山谷,另一种是降低障碍(山坡)。 后者最近才被引入,它的可用性和好处尚未得到充分研究。 这种方法通常被称为加窗 aMD (w-aMD)。 在本教程中,我们将使用原始的 aMD 公式,但是这两个版本现在都可以在 AMBER 上使用。基础aMD引擎对势能的修改被以下方程定义:
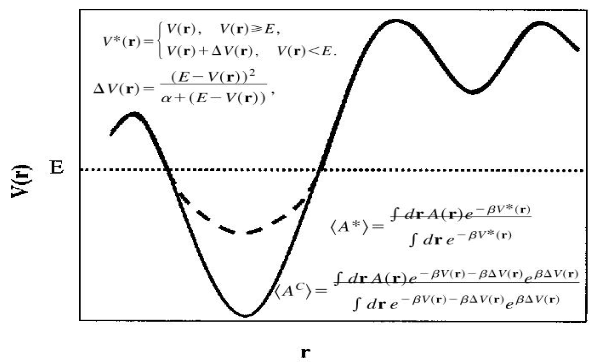
当aMD在AMBER中的实施既可以lowering barriers,又可以raising minima。aMD可以在AMBER的所有MD引擎(sander,pmemd和pmemd.cuda)中实施。The implementation includes the possibility of boosting independently only the torsional terms of the potential (iamd=2) or the whole potential at once (iamd=1). It also allows the possibility to boost the whole potential with an extra boost to the torsions(iamd=3). (目前没搞懂这句的意思)。在aMD中唯一额外需要用户给出的参数是:
a) EthreshP. Average total potential energy threshold. 平均总势能阈值
b) alphaP. Inverse strength boost factor for the total potential energy.总势能的逆强度提升因子
c) EthreshD. Average dihedral energy threshold. 平均二面角能量阈值
d) alphaD. Inverse strength boost factor for the dihedral energy. 二面角能量的逆强度提升因子
本教程将介绍使用aMD的raising minima的采样能力来研究BPTI在微秒时间尺度上发生的构象转变,与毫秒的传统分子动力学模拟进行比较。
Section 1
1) Generating and Relaxing the Initial Structure
普通的MD,将体系平衡。
- Minimize only the water, restraining the protein (20000 cycles)
- Let water move (NTP, 300K), restraining the protein
- Minimize water and protein (20000 cycles)
- Heat the system, restraining the protein (NVT 0 to 300K)
- Relax the system, restraining the protein heavy atoms (NPT, 300K, 0.5ns)
- Relax the system (NPT, 300K, 5ns)
2) Running the aMD calculations and data collection.
有了一个平衡的初始结构后,首先需要运行一个简短的常规MD模拟以收集必要的信息来设置必要的aMD参数。
NSTEP = 39000 TIME(PS) = 6078.000 TEMP(K) = 299.91 PRESS = 94.7
Etot = -39262.0762 EKtot = 7879.4131 EPtot = -47141.4893
BOND = 183.1676 ANGLE = 426.5374 DIHED = 594.3544
1-4 NB = 204.0621 1-4 EEL = 1794.3799 VDWAALS = 7733.9917
EELEC = -58077.9825 EHBOND = 0.0000 RESTRAINT = 0.0000
EKCMT = 3596.2487 VIRIAL = 3332.2057 VOLUME = 129191.4706
Density = 1.0200上述结果只是展示某一帧的能量结果。比如,我们从5ns的MD中获得了 -47,128 kcal/mol 的平均总势能(EPtot)和 595 kcal/mol 的平均二面角(DIHED)能量。 使用这些信息,并考虑到 BPTI 体系有 58 个残基,整个系统 18,226 个原子,我们可以计算 aMD 参数如下:
a) EthreshP: E(tot)= -47128 kcal mol-1 + (0.16kcal*mol-1 atom-1 * 18,226 atoms) = -44212 kcal mol-1
b) alphaP: Alpha(tot)= (0.16kcal mol-1 atom-1 * 18,226 atoms) = 2916 kcal mol-1
c) EthreshD: E(dih)=595 kcal mol-1 + (4kcal mol-1 residue-1 * 58 solute residues) = 827 kcal mol-1
d) alphaD: Alpha(dih)=(1/5)*(4kcal mol-1 residues-1 * 58 solute residues) = 46.4 kcal mol-1
我们现在可以运行完整的 500 ns 模拟,如下所示:
500 ps NVT production NVT &cntrl imin=0,irest=1,ntx=5, nstlim=250000000,dt=0.002, ntc=2,ntf=2,ig=-1, cut=10.0, ntb=1, ntp=0, ntpr=1000, ntwx=1000, ntt=3, gamma_ln=2.0, temp0=300.0,ioutfm=1,iwrap=1, iamd=3, ethreshd=827, alphad=46.4, ethreshp=-44212, alphap=2916, /
pmemd.cuda -O -i amd.in -o amd.out -p ../*.prmtop -c ../eq.rst -r amd.rst -x prod.nc
3) Analyzing the aMD results
未详细看……
4) Reweighting the aMD results
未详细看……
可以重新加权在模拟中获得的分布,以获得未受干扰的分布。
本文来自博客园,作者:计算之道,转载请注明原文链接:https://www.cnblogs.com/jszd/p/16196692.html

 浙公网安备 33010602011771号
浙公网安备 33010602011771号