Improving gene annotation of peanut genome by integrated proteogenomics workflow(通过整合的蛋白质组学工作流程改善花生基因组的基因注释)解读人:卜繁宇
期刊名:J. Proteome Res.
发表时间:2020年5月
IF:3.86
单位:广东省农业科学院
物种:花生
技术:蛋白质基因组学
一、 概述:)
本研究采用蛋白质基因组学策略,整合花生多个组织(种子、壳、雌蕊柄)的RNA-Seq和蛋白质组学数据,以改善花生的基因注释。最终,在蛋白水平上发现了13767个带注释的基因、35个经过修正的基因注释和7个新的蛋白质编码基因。
二、 研究背景:
花生是热带半干旱和亚热带的重要农作物。为了更好地了解花生的分子基础,获得高质量的基因组注释是至关重要的。由于已经对花生基因组进行了测序,因此通过整合基因预测算法、cDNA序列和比较基因组,对花生进行基因注释。但目前的基因注释还不完整。因此,本文旨在通过蛋白质基因组学的方法,整合基因组、转录组和蛋白质组数据,提高花生基因组的基因注释。
三、实验设计:

四、研究成果:
1、新肽的鉴定
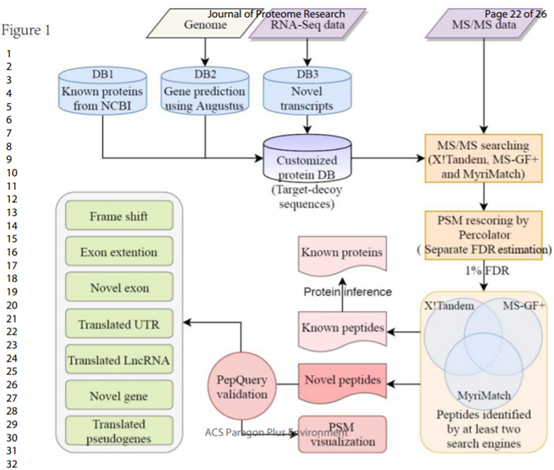
本研究中蛋白质基因组工作流程的概述如图1所示。
自定义蛋白质数据库包括三组不同的蛋白质序列:(1)已知的NCBI蛋白序列(100,775条),(2)利用Augustus基因从头预测的蛋白序列(698,820条)和(3)RNA-Seq数据衍生的新转录本蛋白序列(967,048条),去除冗余序列,最终得到1,692,402条蛋白序列。
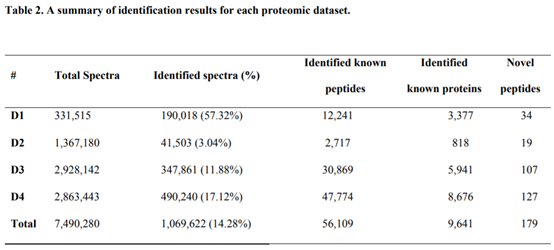
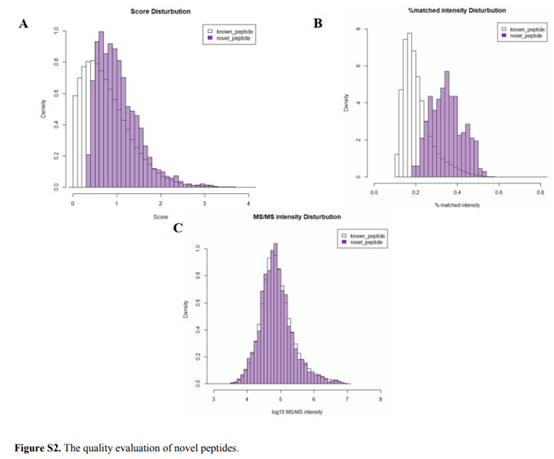
三个搜索引擎(MS-GF+, X!Tandem and MyriMatch)比对自定义蛋白质数据库,共鉴定出56,288个肽段(表2总结了每个数据集的详细鉴定结果)。其中,56,109个已知的肽段可以比对到已知的蛋白上,179个新肽段不能比对到任何已知的蛋白序列上。这179个新肽段中,有122个(68.2%)同时存在于DB2和DB3中,33个(18.4%)只存在于DB2中,24个(13.4%)只存在于DB3中。并且这些新肽的质量较高(见图S2)。
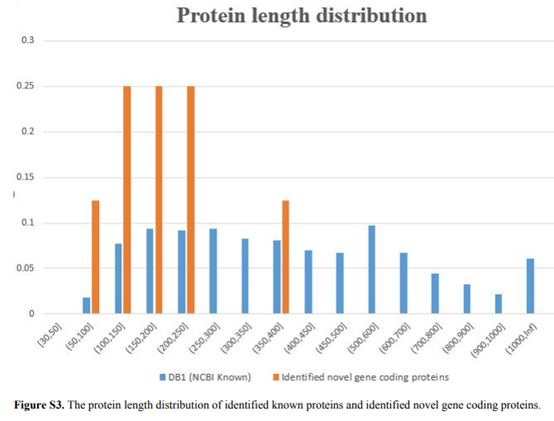
如图S3所示,新鉴定蛋白质的平均长度为198,已知蛋白质的平均长度为456。新蛋白质的平均长度比已知蛋白质短,这可能表明编码短蛋白的基因在之前的注释中容易丢失。
2、对现有注释的验证
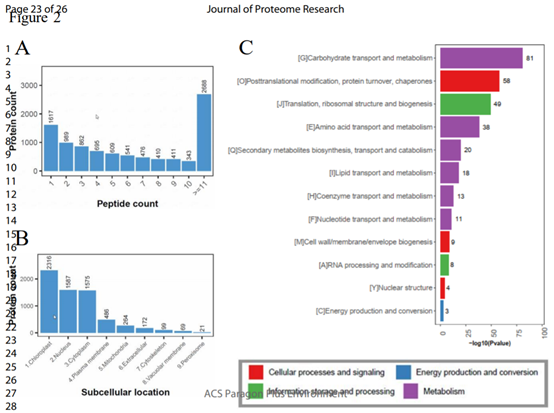
如图2所示,本研究共鉴定9,641个蛋白组(17,037个蛋白),其中8024个蛋白(83%)具有两个以上unique肽段,鉴定结果高可信度;其中70%的蛋白位于叶绿体、细胞核和细胞质上。此外,这些鉴定的已知蛋白主要参与生长、发育和代谢途径。
3、修正注释基因模型
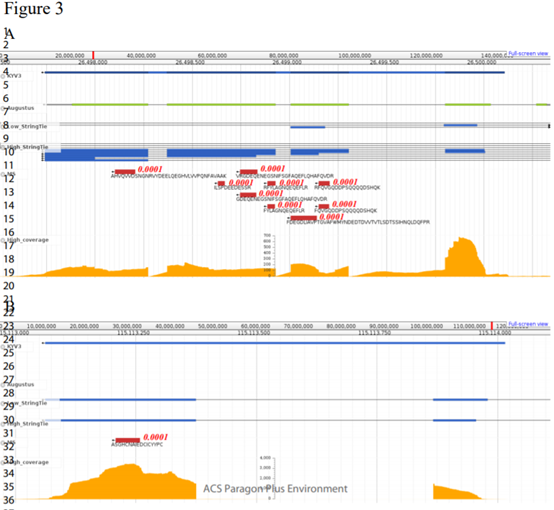
本研究通过对自定义蛋白质数据库的MS/MS数据搜索,检测到35个修订的基因。如表3所示,本研究找到了25个假基因的翻译证据,几个新的肽段被唯一的比对到2个LncRNA上。通过BLAST进一步验证这些假基因的编码可行性,结果显示这些蛋白与已知蛋白具有较强的序列相似性(blast E-value < 1e-30)。功能分析表明,这些LncRNA具有与RNA加工修饰相关的功能。图3显示了可翻译的假基因和可翻译的LncRNA。
4、新基因
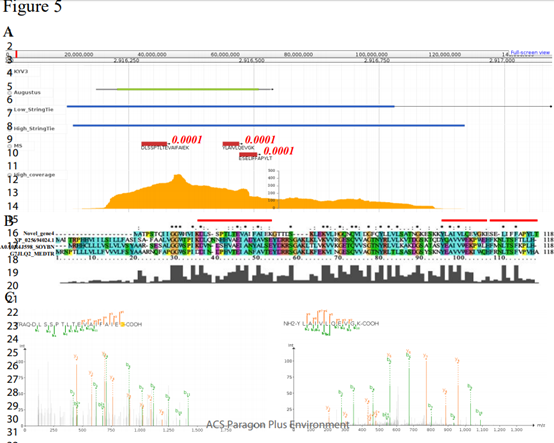
本研究使用Augustus来预测潜在的新基因模型并根据RNA-Seq数据预测新的转录本,两种方法分别预测了524,202和40,815个潜在基因。在本研究中共鉴定了7个新的蛋白质编码基因,这些基因具有≥2个肽段,其中至少一个是unique肽段。共有17个新肽段被定位到7个新基因上。利用BLAST比对NCBI非冗余数据库,结果显示这些新基因与至少一种已知蛋白质具有显著的序列相似性。根据其序列相似性,利用Blast2GO和KOBAS推断新基因的功能,发现几个新的基因匹配到ATP合酶β亚基。图5展示了一个新基因。
五、文章亮点(结论讨论):
采用蛋白质基因组学策略,整合RNA-Seq和蛋白质组学数据来改善花生的基因注释,提高了对花生生物学特性的理解,特别是7个新的蛋白质编码基因和35个修订的基因注释。此外,本研究中使用的研究方法可用于对其他物种进行蛋白质组学分析。
阅读人:卜繁宇
DOI:10.1021/acs.jproteome.9b00723

 浙公网安备 33010602011771号
浙公网安备 33010602011771号