Detergent-Free Simultaneous Sample Preparation Method for Proteomics and Metabolomics (一种无变性剂的同时制备蛋白质组和代谢组样品的方法)
文献名:Detergent-Free Simultaneous Sample Preparation Method for
Proteomics and Metabolomics (一种无变性剂的同时制备蛋白质组和代谢组样品的方法)
期刊名:Journal of proteome research
发表时间:(2019年11月)
IF:3.8
单位:
- 圣詹姆斯大学医院
- 利兹大学
样本:肾癌组织活检样本
技术:多组学研究样本处理,无变性剂样本处理,SiTrap
一、 概述:
组学技术的整合逐步改变我们对生物系统的理解。但是当样本量有限时,独立的样本处理方法影响了多组学的研究。该研究描述了一种简单、有效的无变性剂同时富集的样本处理方法,有利于同时测定同一样本提取物中的蛋白组和代谢组。这种一次性的多组学处理过程有利于对样品数量有限或者异质性的样本的处理,如组织活检样本。本文利用SiTrap技术对肾脏癌组织进行检测,展示该方法作为一种重要的多组学工具的价值。
二、 研究背景:
组学技术的结合加深了我们对生物系统和人类病理学的理解。然而,跨组学平台的整合分析带来了新的挑战。组学的样本制备技术通常依赖平台并相互独立。我们对组学的研究受限于技术中的破坏性,由于样本被降解以提取DNA、RNA、蛋白质、代谢物或脂质。这对微量样本和异质性样本尤其不利,当多次样本处理不实际或者产生矛盾的结果。该研究团队之前发表了STrap蛋白质组学样本处理相关的方法。该方法将SDS变性和蛋白烷基化,过滤去除SDS和污染物后酶解集成在Strap管中,方便、快捷。但是在这个过程中,存在一个问题,如果下游的组学是全谱的,例如代谢组学,流穿的部分需要收集,然而里面含有的变性剂、还原剂、烷基化试剂都会干扰代谢组学的分析。目前,同时提取的方法很少被报道。已报道的方法基于相分离,例如氯仿甲醇萃取方法,受其复杂性且耗时的限制,并且在样品量少及高通量分析的情况下均不实用。因此,本文开发了与STrap的蛋白处理相匹配的方法,但是使用无变性剂进行裂解,并对富集后的蛋白进行原位还原和烷基化,为补充的组学分析提供无污染的流穿组分。
三、实验设计:
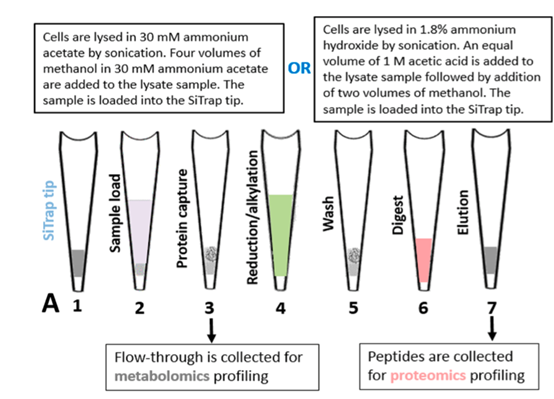
图A. 实验策略
四、研究成果:(重点图表展示)
1、为了测试新的SiTrap方法的蛋白质组学性能,该研究通过提取MDA-MB-231细胞的蛋白将它与STrap方法进行了比较,结果使用AA和AH进行裂解的SiTrap方法中,分别鉴定到1293 (±12 SD) 和1278 (±44 SD) 个蛋白,在用SDS裂解的STrap方法中鉴定到1230 (±27 SD)个蛋白(图1B)。在主要GO细胞组分分类中蛋白的分布在3种方法下都非常类似(图1C),并且大多数蛋白通过三种方法都可以被鉴定到,没有偏向性(图1D)。
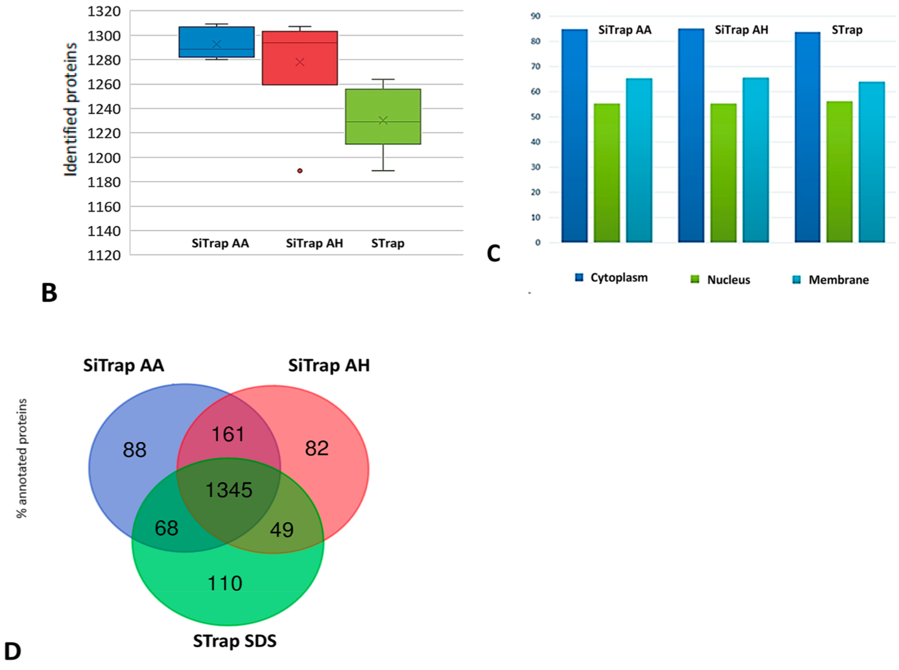
图1.(B)鉴定蛋白数目的箱形图(一个蛋白中至少有2个肽段被鉴定到);(C)在主要的细胞组分中蛋白的分布;(D)3组蛋白制备方法中鉴定到蛋白的韦恩图;
2、利用SiTrap对肾癌和癌旁组织进行分析,鉴定到2655个蛋白,62种代谢物。这其中59个代谢物可被定量,包括25种游离脂肪酸、19种酰基肉碱和15种胆汁酸。通过对这些代谢物进行分析,在癌症样本中,短链丙酰丙氨酸(C5、C5:1和C3)和多重不饱和脂肪酸(C20:5、C20:4、C22:6)均有减少(图2A,图3A)。肉碱O-乙酰转移酶(CRAT)、肉碱O-棕榈转移酶2(CPT2)和肉碱O-棕榈转移酶1(CPT1A),这些在丙酰胺代谢中具有关键作用的酶,被鉴定和定量,并发现在癌症样本中显著减少,着与代谢组结果一致(图2B,图3B)。
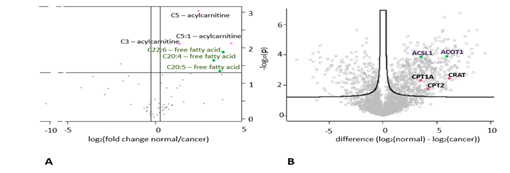
图2.癌症和癌旁组织代谢组和蛋白组的火山图显著性分析。
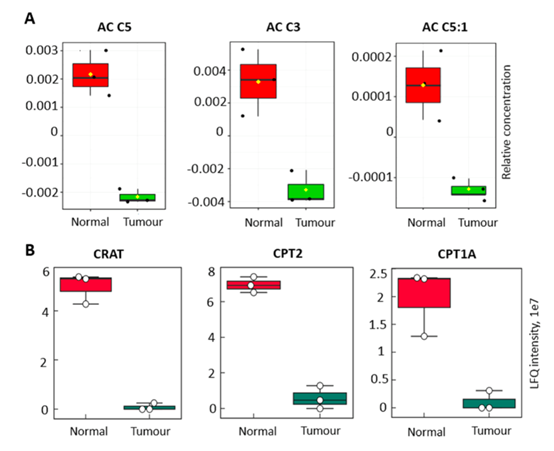
图3. SiTrap方法中肾癌组织的蛋白组和代谢组中鉴定到的酰基肉碱代谢紊乱相关代谢物和酶。
五、文章亮点:
该研究概述用于蛋白质组和代谢组无变性剂样本处理的同步富集方法SiTrap。
该方法为在同一样本中进行简单而可靠的多组学分析提供了机会。
阅读人:陈凌云

 浙公网安备 33010602011771号
浙公网安备 33010602011771号