Journal of Proteome Research | iHPDM: In Silico Human Proteome Digestion Map with Proteolytic Peptide Analysis and Graphical Visualizations(iHPDM: 人类蛋白质组理论酶解图谱的水解肽段分析和可视化展示)| (解读人:邓亚美)
文献名:iHPDM: In Silico Human Proteome Digestion Map with Proteolytic Peptide Analysis and Graphical Visualizations
期刊名:Journal of Proteome Research
发表时间:(2019年12月)
IF:3.86
单位:
- 资讯科学研究院,中央研究院,台湾
物种:人
技术:蛋白质组生物信息学
一、 概述:(用精炼的语言描述文章的整体思路及结果)
在有关丢失蛋白(MP,missing protein)和蛋白质亚型检测的蛋白质组学研究中,蛋白水解酶的选择是实验设计时需要考虑的一个因素。为了便于蛋白水解酶的选择,本研究开发了iHPDM的网页工具,用于计算机模拟蛋白质消化过程和结果展示。在进行鸟枪法蛋白质组学实验时,iHPDM能够指导蛋白水解酶的选取,以便鉴定MPs、蛋白质亚型和单氨基酸突变肽段。
二、 研究背景:
现阶段,寻找丢失蛋白、注释蛋白质及其亚型是人类染色体蛋白质组计划(C-HPP)的两大主要目标。据统计,neXtProt数据库(2019年1月版本)收纳了20339个具有代表性的人类蛋白质,其中MPs仍有2129个。一直以来,以肽段分析为核心的鸟枪法(shotgun)蛋白质组学是鉴定MPs和蛋白质亚型的常用策略。该策略下蛋白质先经蛋白酶水解为肽段,水解后的肽段经由液相色谱分离并进行质谱检测,其中,蛋白水解酶的选择将会影响可检测肽段的种类和数目,进而限制蛋白质鉴定和蛋白质序列覆盖度。如何合理地选择蛋白水解酶是该研究关心的核心问题。
胰蛋白酶是一种最常见的蛋白水解酶,水解后肽段的质量区间近似在0.5~3 kDa,适用于鸟枪法蛋白质组学的实验流程,使用率高达96.3%。然而,胰蛋白酶对蛋白质切割活动具有一定的阻碍效应(hinder effects),这会导致蛋白质消化不完全以及漏切位点的存在,不利于MPs的鉴定。为了减少此类限制,在蛋白质组学实验中使用其他类型蛋白酶或者联合使用多种蛋白酶将是蛋白质消化的替代解决方案。这些方案有望产生更多unique肽段和更高的序列覆盖度,不仅利于MPs和蛋白质亚型的鉴定,还能够为单氨基酸变体(SAV)肽段的鉴定提供更多可检测的肽段。
为了选择合适的蛋白酶,研究人员需要借助计算机模拟蛋白质消化过程,并通过对模拟消化肽段的结果分析,设计最佳的蛋白质组学实验。目前,已经存在一些Web服务器和独立工具可用于这方面的分析,如PeptideCutter,IPEP,PeptideMass,Proteogest,PepServe和PeptideManager。但是,这些工具尚存在一些不足,主要表现在4个方面:1)展示的结果信息不够全面。如PeptideCutter和IPEP缺乏关于肽段序列唯一性和长度的信息;2)缺乏蛋白质亚型检测的功能。针对目标蛋白质亚型的检测,缺少关于蛋白酶组合适配性方面的检测分析;3)不支持对查询蛋白的消化肽段进行灵活的动态过滤;4)缺乏蛋白质消化结果的可视化审阅功能。
鉴于现有软件的不足之处,该研究构建了一个新型网络服务器工具以满足需要,并命名为iHPDM(in silico Human Proteome Digestion Map),使用地址为http://ms.iis.sinica.edu.tw/iHPDM/index.php。 iHPDM功能全面,其专业化的蛋白酶推荐功能将能够更好地指导蛋白质组学实验,推进丢失蛋白、蛋白质亚型和变异肽段鉴定的研究。
三、 iHPDM数据库资源的构建与功能
为了开发人类蛋白质组的蛋白质酶解消化图谱,iHPDM使用了neXtProt数据库中所有人类蛋白质(2019-01版,包括蛋白质亚型在内的42419个蛋白质序列)作为数据源;提取了每个蛋白的身份和属性等信息(包括neXtProt索引号、蛋白质名称、蛋白质长度、染色体位置等);支持一种或两种蛋白酶的15个组合对42419个人类蛋白质进行计算机消化(酶组合:trypsin,chymotrypsin,LysargiNase,ArgC,GluC,LysC,LysN,AspN,OmpT,KEX2,SAP9,LysC + AspN,trypsin + GluC,trypsin + LysN和trypsin + AspN)。

图1 iHPDM的主界面
从图1 iHPDM的主界面来看,它提供了3大功能模块:
(1)Protein Query模块:针对单一蛋白序列,支持15种组合酶的平行比较分析。 示例结果如图2所示。
(2)Multi-protease Comparison模块:适用于高达1000条蛋白序列的批量处理,支持至多5种组合酶的水解效率的比较评估。
(3)Isoform Digestion模块:在给定蛋白质名称和蛋白酶的情况下,提供了不同蛋白质亚型经蛋白酶消化后地图形可视化结果展示,以便选择最适蛋白酶。示例结果如图3所示。

图2 关于NX_Q14390蛋白的Protein Query模块操作结果演示。
在蛋白质序列视图中,所有消化所得肽段按分子量大小归类为BUP(bottom-up proteomics,0.6-3 kDa),eBUP(extended bottom-up proteomics,3-7 kDa)或MDP(middle-down proteomics,7 -15 kDa)三类。
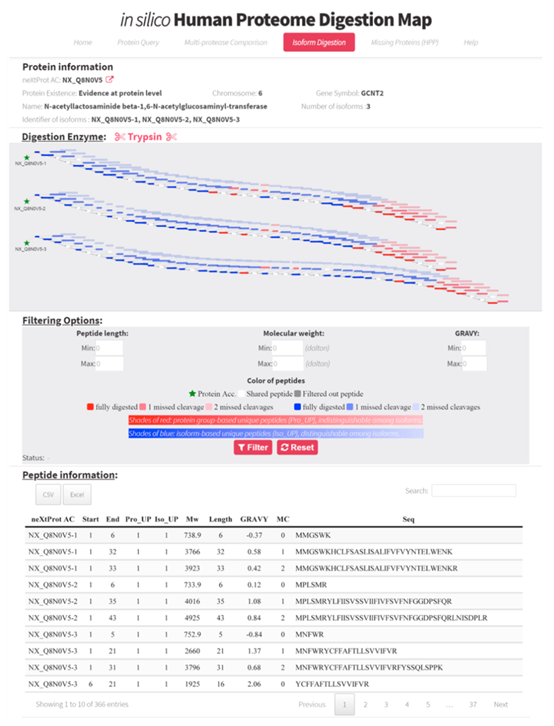
图3 胰蛋白酶作用下三种NX_Q8N0V5蛋白质亚型的消化结果展示
五、文章亮点(结论讨论):
本文提供了一个功能全面的、可对蛋白质进行理论酶解的iHPDM网页版工具。该工具的亮点是1)支持蛋白酶的种类丰富;2)提供了交互式图形操作和可视化界面,便于分析和检查消化结果;3)不仅可以鉴定MPs和蛋白质亚型,还可以选择蛋白酶以便检测具有单氨基酸变异肽的蛋白质。
阅读人:邓亚美
原文链接:https://pubs.acs.org/doi/abs/10.1021/acs.jproteome.9b00350

 浙公网安备 33010602011771号
浙公网安备 33010602011771号