
Mol Cell Proteomics. | MARMoSET – Extracting Publication-ready Mass Spectrometry Metadata from RAW Files
本文是马克思普朗克心肺研究所的三名研究者Marina Kiweler、Mario Looso和Johannes Graumann发表在8月刊的MCP的一篇文章。
由于Omics实验经常涉及数百个数据文件,元数据信息对于结果的评估和再现至关重要;然而数据通常以二进制或专有文件格式存在,元数据信息提取过程繁琐。以Thermo Fischer Scientific质谱仪生产的RAW文件为例,除了光谱数据之外,还包括了仪器设置,这都是实验评估和结果再现所必需的。目前提取RAW文件信息的方法是使用特定供应商的Xcalibur软件手动打开RAW文件并且复制所需信息,然而手动提取容易出错并且存在访问量过大的问题。迄今为止没有软件能够解决将大量RAW文件或其他元数据简化成具有共识参数的报告的问题。通过供应商提供的RAWFileReader应用程序的API接口,作者基于R的基础架构编写了一个工具,可以从元数据中提取并生成用于实验室质量控制的数据报告。

图1 MARMoSET处理过程概览
MARMoSET分为两部分,第一部分是C#应用程序,它的作用是从Thermo Fischer Scientific RAW文件中提取元数据信息为JSON文件。通过RAWFileReader API访问的RAW文件格式包含了多个层次的元数据信息。固定标题包括日期,原始文件名和样本信息等信息。标题后面是一个列表,其中包含使用的仪器模块以及它们各自的方法作为字符串。API还为检测器相关数据(如紫外分光光度法或质谱法)提供单独的入口点。MARMoSET目前仅实现对MS数据的访问。使用RAWFileReader API中的“IRAW DataPlus”接口,在使用EASY-nLC超高压液相色谱仪器(Thermo Fisher Scientific)的液相色谱/质谱(LC / MS)的背景下,LC参数可在方法串中获得,并由MARMoSET提取和分析。根据提供的是单个RAW文件还是文件目录的路径,MARMoSET可以判断作用于单个文件或迭代目录中的RAW文件集合,并根据计算机的硬件资源作并行化处理。在第一步中,分别从每个RAW文件收集信息。第二步,为了将来自多个文件的数据减少为描述整个集合的最小参数集,所得到的数据结构是在字典中评估和排序过后的哈希码。然后,此信息用于将RAW文件分类为可以共享所有相关参数的组。最后编写成可以连接到相应RAW文件名的JSON文件。为了方便直观地处理JSON文件中的结构化数据,作者又同时提供了一个名为MARMoSET的R包。它能够根据预先定义的日志文件创建表格,此外还支持通过单独选择参数来过滤数据。
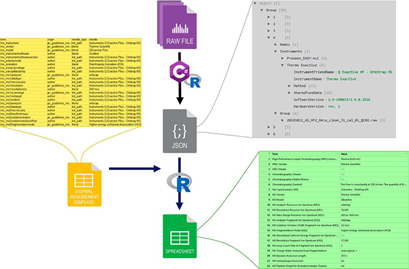
图2 MARMoSET处理过程
在windows操作系统上,通过直接运行C#命令行工具,可以生成JSON文件,基于R包中自带的术语匹配表,使用函数“match_terms()”可以提取对应参数的子集并生成日志特定要求的表格,然后使用函数“save_all_groups”导出表格。元数据的标准化报告对于实验的评估和再现极为重要,MARMoSET工具套件的诞生填补了其空白,生成了面向机器可读的JSON文件和面向人类可读的txt或excel文件。
一方面解决了Omics实验高吞吐量元数据的处理问题,另一方面R包所提供的自定义参数设置可以灵活满足不同实验的要求,过滤不必要的信息。
MARMoSET C#应用程序:https://github.molgen.mpg.de/loosolab/MARMoSET_C
MARMoSET R包:https://github.molgen.mpg.de/loosolab/MARMoSET
解读人:马臻
文章引用:10.1074/mcp.TIR119.001505
文章连接:https://www.mcponline.org/content/18/8/1700

 浙公网安备 33010602011771号
浙公网安备 33010602011771号