Mol Cell Proteomics. | Integration and analysis of CPTAC proteomics data in the context of cancer genomics in the cBioPortal (解读人:徐洪凯)
文献名:Integration and analysis of CPTAC proteomics data in the context of cancer genomics in the cBioPortal
期刊名:Molecular & Cellular Proteomics
发表时间:2019年9月
IF:4.828
作者:
Pamela Wu1,2,3, Zachary J Heins4, James T Muller3, Lizabeth Katsnelson3, Ino de Bruijn4, Adam A Abeshouse4, Nikolaus Schultz4,5, David Fenyö1,2, Jianjiong Gao4,5
单位:
- 纽约大学医学院,生物化学与药理学系,纽约10016
- 纽约大学医学院,系统遗传研究所,纽约10016
- 纽约大学医学院,萨克勒研究所,纽约10016
- 纪念斯隆-凯特琳癌症中心,Marie-Josee和Henry R. Kravis分子肿瘤学中心,纽约10065
- 纪念斯隆-凯特琳癌症中心,流行病学和生物统计学系,纽约10065
物种:人(Homo sapiens)
技术:cBioPortal
一、 概述:
临床蛋白质组学肿瘤分析联盟(Clinical Proteomic Tumor Analysis Consortium, CPTAC)旨在通过大规模蛋白质组和基因组分析(或蛋白质基因组学)的应用,促进人们对癌症分子基础的理解。cBioPortal(http://www.cbioportal.org/)是一个能够对多维癌症基因组和临床数据进行挖掘、可视化和分析的开源平台。本文中开发了用于把由CPTAC成员产生质谱结果到的数据格式与cBioPortal数据管道兼容的数据转换流程。并将CPTAC中的77例乳腺癌、95例结肠癌和174例卵巢癌的蛋白质组学数据整合到了cBioPortal中,以便于在相同肿瘤的基因组和临床数据背景下,对蛋白质组数据进行综合分析。结合TCGA中的基因突变、拷贝数改变、基因表达和DNA甲基化信息,整合以后的CPTAC数据可以比较容易地在cBioPortal的基因组和临床信息背景下进行分析挖掘。
二、 研究背景:
过去十年中,TCGA(The Cancer Genome Atlas)发布了三十多种肿瘤的基因组数据,并利用反相蛋白阵列平台(the Reverse Phase Protein Array, RPPA)产出了蛋白质组数据,以评估肿瘤中将近150个蛋白质和50个磷酸化蛋白的丰度水平。然而RPPA技术受蛋白质和翻译后修饰的检测抗体的有效性和结合效率限制,已经不能满足当前的需求。目前,CPTAC所延伸的蛋白基因组学(Proteogenomic)技术在已经在分析包括透明细胞肾细胞癌、子宫内膜癌、肺腺癌、肺鳞状细胞癌、胶质母细胞瘤、头和颈部鳞状细胞癌和胰腺导管腺癌等其它类型癌症中得到了应用。通过对TCGA中已经分析过的同一癌症患者进行分析,CPTAC的为癌症基因组学和蛋白质组学数据的关联分析提供了新的角度,并将蛋白质组学与癌症的潜在的表型联系起来。cBioPortal是癌症基因组学研究中重要的资源之一,它包括所有TCGA项目中的数据集以及从文献中整理的多个数据集,加上它所具有的用户友好界面、可视化和综合分析功能,使得cBioPortal成为癌症基因组学研究人员中最受欢迎的资源之一。

图1. cBioPortal 功能接口介绍。
三、 实验设计:
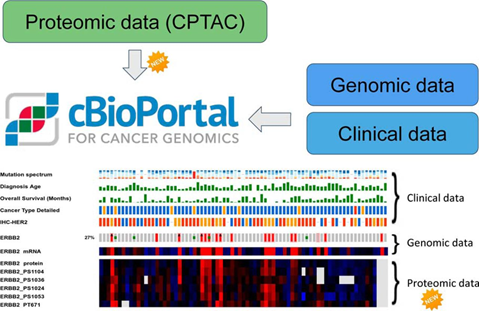
图2. 肿瘤临床信息、基因组和蛋白质组数据的整合示例
四、研究成果:
1. CPTAC 中的数据利用CDAP(Common Data Analysis Pipeline)流程进行标准化,然后利用MS-GF+进行肽段鉴定。
2. 根据RefSeq ID将蛋白质匹配到相应的基因中,并利用特异肽段进行定量。
3. 对翻译后修饰(主要指磷酸化)添加特殊标签进行区分:<HUGO gene symbol>_P{S,T,Y}<AA modified>。例如,在位于64位丝氨酸上磷酸化的EIF4EBP1表示为EIF4EBP1_PS65。同时为磷酸化蛋白定义别名PHOSPHO<HUGO gene symbol>便于查询。
4. 将质谱信号强度转换为每个细胞蛋白质拷贝数的估计值(可选功能):
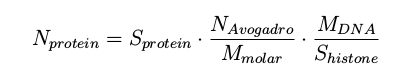
N代表数量(阿伏伽德罗数,6.022*1023,和所测量的蛋白质的拷贝数),S代表质谱信号值(组蛋白和所测得的蛋白质),M代表每个细胞DNA的质量(估计值为6.5 pg和测量的蛋白质的摩尔质量)。
5. 支持来自CDAP和MaxQuant中产生的蛋白质组和PTM分类文件。
目前cBioPortal 已经有了诸多应用:Web界面有助于探究癌症患者的mRNA和蛋白质表达数据的调控模式;使用R包cgdsr通过其web API探索cBioPortal数据,以访问现在加载了质谱数据的完整数据库;以及加载未公布的或特定机构的蛋白质组学数据到本地私人cBioPortal实例的综合分析。
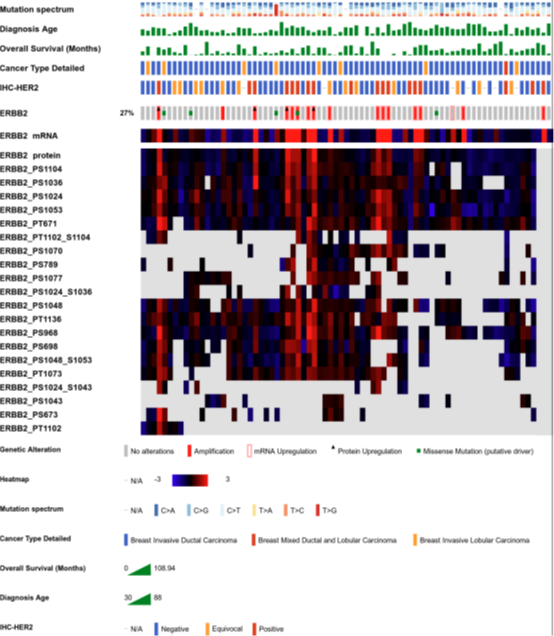
图3. 来自TCGA的77例乳腺癌的临床信息、基因组和蛋白质组特征的OncoPrint图形展示。
示例中的临床信息包括突变谱、诊断年龄、总生存期、详细癌症类型和HER2(ERBB2)IHC分数,免疫组化(immunohistochemistry)打分用于说明说明免疫组化染色是否能够检测到肿瘤表面的ERBB2受体:阴性为0-1分,分辨模糊为2分,阳性为3+。ERBB2基因组和蛋白质组的可视化特征包括基于CPTAC数据的基因突变、拷贝数扩增、mRNA、蛋白质和蛋白质磷酸化水平等。
五、文章亮点:
用于进行交互式综合信息分析的cBioPortal为应对高通量测序和质谱技术发展所带来的整合多个组学信息的挑战提供了有力的帮助。将CPTAC中的质谱数据整合到cBioPortal中,提高了基因组学的背景下基于质谱技术的蛋白质组学数据的可访问性,并为癌症研究人员探索和分析癌症基因组学和蛋白质组学之间的相互作用提供了一个直观的界面。目前基于质谱技术分析的肿瘤样本的数量仍远小于反相蛋白阵列分析的样本量(表1),预计在未来基于质谱技术的肿瘤研究将会大大增加。将质谱数据与基因组和临床数据整合,将在揭示癌症基因组、蛋白质组和表型之间的新的关联提供了机会。
表1. cBioPortal中使用CPTAC质谱数据进行的癌症研究,截至2019年7月。
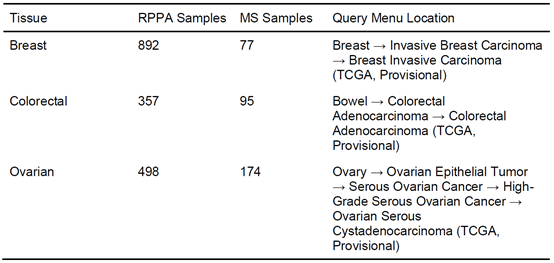
注:Query Menu Location是指在cBioPortal主页上组织癌症研究的树状结构中设置初始查询参数的位置。
阅读人:徐洪凯
原文链接:https://www.mcponline.org/content/18/9/1893
DOI:https://doi.org/10.1074/mcp.TIR119.001673

 浙公网安备 33010602011771号
浙公网安备 33010602011771号