Journal of Proteome Research | Clinically Applicable Deep Learning Algorithm Using Quantitative Proteomic Data (分享人:翁海玉)
题目:Clinically Applicable Deep Learning Algorithm Using Quantitative Proteomic Data
期刊:Journal of Proteome Research
发表时间:August 2, 2019
DOI:: 10.1021/acs.jproteome.9b00268
分享人:翁海玉
内容与观点:
本研究描述了一种优化的基于深度学习(DL)的胰腺癌诊断方法并测试了该方法的分类能力。
1、实验设计
1.1数据集构建:该方法使用1008个样本的选择反应监测-质谱(SRM - MS)数据集,SRM-MS在血浆样本中检测出34种多肽(由34个蛋白衍生而来)。数据集包括300个正常人样本(NC),109个胰腺癌良性样本(PB),49个其他良性样本(OB),149个其他癌症样本(OC),和401个胰腺癌样本(PDAC)。按照0.7:0.3的比例将数据集分为训练集(691 samples; 322 PDAC, 41 OB, 88 PB, and 240 NC)和测试集(317 samples; 79 PDAC, 8 OB, 149 OC, 21 PB, and 60 NC),保持内部比例不变。其中OC只在测试集中有,以确定是否构建的模型会受到癌症异质性影响。
为了算法能够表现出鉴别胰腺癌的能力,数据集被重新构建为控制组(NC+PB+OB+OC),病例组(PDAC)。
1.2 DL模型训练和参数优化:采用十倍交叉验证的方法对训练数据集进行处理,避免了抽样偏差。每次迭代从子训练数据集中随机抽取约622个数据点(691*0.9)输入模型;其余69个值(691*0.1)作为子测试数据集,用于评估模型中的误差,同时对每个选定的数据点(分层抽样)保持对照组和病例组的比例相等。为了构造该模型,我们采用逐步逼近的方法来减少测试所有可能特征集的计算量。
利用训练数据集对模型进行微调,优化参数。然后在独立的测试数据集上对训练后的模型进行测试,并对其分类性能进行评估。利用独立的测试数据集进一步验证了模型的性能。利用测试数据集的性能来指导参数的优化。为了减少样本选择偏差和模型过拟合的可能性,除了交叉验证外,还进行了bootstrapping验证。
训练和测试数据集使用v3.10.3.6版本的H2O软件包进行处理。DL方法对10个最重要的参数纪元数(number of epochs)、节点数和隐层数(number of nodes and hidden layers)、激活函数(activation function)、rho、epsilon、L1 & L2正则化(L1 & L2 regularization)、隐藏丢失率(hidden dropout ratio)、输入丢失率(input dropout ratio)、每次迭代训练样本(train samples per iteration)、最大w2(max w2)。同时进行网格搜索来优化每个参数的值。并使用每个参数的常用值对它们逐一进行了优化,以此确定重要参数。
1.3 五种传统机器学习模型参数优化:对在蛋白质组学应用最广泛的五种机器学习模型:随机森林(RF)、支持向量机(SVM),逻辑回归(LR),K近邻(KNN)和贝叶斯(NB)建模,训练和测试数据集的处理与DL方法相同。用网格搜索,对5种方法中的参数进行调优。
1.4 DL与传统模型比较:
采用了五种传统的模型性能指标:查全率、精密度、F1评分、精密度和工作特性曲线下面积(AUROC):
Recall= 
Precision= 
F1 score= 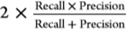
Accuracy= 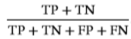
AUROC通过测量这个图的recall和FDR来构建AUROC曲线,其中1.0表示完全分离,0.5表示随机分类。如图:
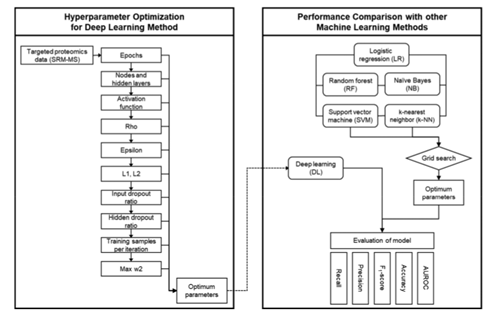
Figure 1 完整实验过程
2、结果
2.1 DL参数优化:10个参数中 epoch, activation function, epsilon, input dropout ratio影响DL模型的分类模型(Figure 2 ),如图,选择了AUROC最大时的值为参数值。
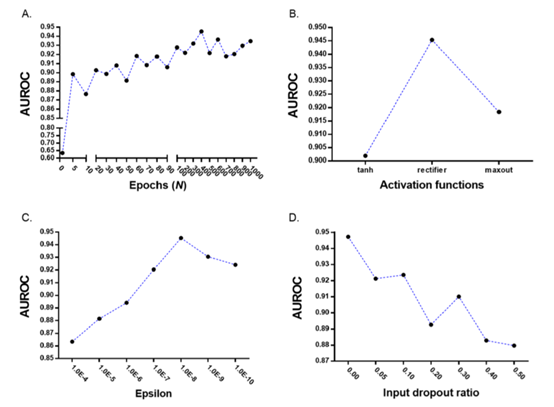
Figure 2 DL参数优化
2.2 DL与传统机器学习模型比较:
各个指标都有明显提升,如下图:
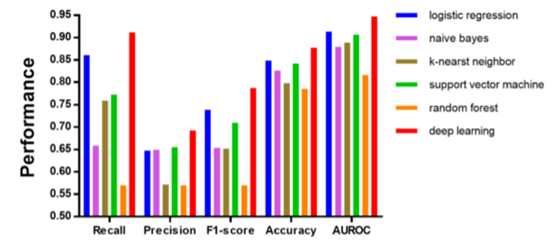
Figure 3 六个模型的性能参数柱状图
3、结论
研究结果表明,DL是蛋白组学数据生物标志物确认的有力工具。在临床实验室中,DL有提高疾病分类任务的标准化和内部可靠性的潜力。未来的工作应该优化其在临床环境中的表现,以充分利用DL方法作为临床工具。
4、讨论
虽然DL各个性能指标都远远高于传统方法,但其仍然存在耗时长,电脑硬件要求高,需要更多的特征和样本的数据集等局限,尤其受到质疑的是,DL是一个黑盒子,难以给出内部过程。但本文向我们展示了DL的潜力。相信DL预测不同群体的高精度的能力将产生全新的数据处理选项,支持和加强未来基于蛋白组学的生物标志物研究。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号