解读人:刘佳维,Spectral Clustering Improves Label-Free Quantification of Low-Abundant Proteins(谱图聚类改善了低丰度蛋白的无标记定量)
发表时间:(2019年4月)
IF:3.95
单位:
- 维也纳医科大学;
- 欧洲生物信息研究所(EMBL-EBI);
- 分子病理学研究所;
- 奥地利科学院分子生物技术研究所;
- Gregor Mendel分子植物生物学研究所。
对象:质谱无标记定量结果
技术:聚类分析
一、 概述:(用精炼的语言描述文章的整体思路及结果)
本文选择四个不同的数据集,分为基于谱图数计数和基于峰值强度计数的无标记定量两种情况,对谱图进行聚类算法分析,提高了低丰度蛋白的可检测性,并开发了可直接使用的聚类方法的PD节点。
二、 研究背景:
无标记量化已成为许多基于质谱的蛋白质组学实验中的常见做法。近年来,聚类方法可以改善蛋白质组学数据集的分析的结论已广泛被人们所接受。本文旨在利用光谱聚类推断额外的肽谱匹配,并提高数据集中的无标记定量蛋白质组学数据的质量,改善低丰度蛋白的定量结果,同时提高了衍生定量数据的准确性,且没有增加数据集的噪声。
三、 实验设计:

图 1:基于谱图计数和基于强度计数两种方法对LFQ进行聚类以得到额外PSMs的工作流程。
名词解释:
LFQ:Label-Free Quantification,无标记定量;
MGF:Mascot genetic format,一种文件格式;
PSMs:peptide spectrum matches,匹配到的肽段谱图;
MSGF+/X!tandem:常用的搜库软件;
MS-Amanda:PD中常用的搜索算法;
apQuant:通过质量过滤使LFQ的结果更准确。
四、研究成果:
1、以在酵母蛋白环境下加入不同浓度的做了标记的UPS1蛋白的样品进行常规蛋白分析得到ms谱图,这些UPS1蛋白即为所用样品中的用来检测结果的低丰度蛋白,然后在搜库时选择是否使用聚类方法并将检测到的标记低丰度蛋白量进行比对,结果如图2。可以看出在低浓度情况下使用聚类方法检测到的低丰度蛋白量提升更明显。
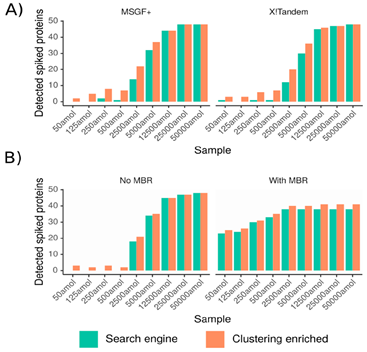
图 2:横坐标为加入的不同摩尔数的标记UPS1蛋白,纵坐标为检测到的标记UPS1蛋白量,并根据是否使用聚类算法将结果表示为橙绿两种颜色。其中:(A)基于谱图数计数,分别使用MSGF+与X!Tandem搜索引擎;(B)基于峰强计数,并分为是否使用MBR(match-between-runs,边运行边匹配)两种情况。
2、将结果蛋白中有标记的视为真阳性,属于背景蛋白的视为假阳性,通过改变判断结果蛋白是否达标的阈值,绘制出聚类方法在不同情况下与常规方法效果的比对图,曲线面积越大说明越能在更低的假阳性率下获得更高的真阳性率。从图中我们可以看出聚类方法在大部分情况下都对结果有所改善。

图 3:分别使用limma对(A,B)和edgeR对(C)做出统计分析,横坐标为假阳性率,纵坐标为真阳性率,线的颜色代表是否使用聚类方法,虚实代表是否使用MBR(A,B)或所用搜索引擎种类(C)。 其中:(A)基于峰强计数,使用三个CPTAC数据集得到的结果。 (B)基于峰强计数,三种浓度比得出结果。(C)基于谱图数计数,三种浓度比得出结果。
文章亮点:
本文最大的亮点在于将其开发的光谱聚类算法整合到了广泛使用的PD软件套件中,使其更容易被更广泛的蛋白质组学界所用。能直接使用的PD节点可在http://ms.imp.ac.at/?goto=spectra-cluster 下载。此外,聚类方法不依赖数据库,但可以直接使用库里的谱图,这使其有着更高的灵敏度。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号