MCP|LQD|Data-independent acquisition improves quantitative cross-linking mass spectrometry (DIA方法可提升交联质谱定量分析)
文献名:Data-independent acquisition improves quantitative cross-linking mass spectrometry (DIA方法可提升定量交联质谱分析)
期刊名:Mol Cell Proteomics
发表时间:(2019年4月)
IF:5.232
单位:
1. 德国柏林技术大学生物技术研究所
2. 英国爱丁堡大学生命科学学院
3. 瑞典 Biognosys 公司
技术:DIA交联质谱定量
一、概述:(用精炼的语言描述文章的整体思路及结果)
建立了用DIA的质谱数据采集和分析方法来对交联蛋白做定量分析的流程,通过比较已知不同浓度的单独交联的七个已知晶体结构的蛋白用定量交联质谱得到的交联肽段定量值,发现基于DIA的定量交联质谱方法比基于DDA的定量交联质谱方法有更高的可定量比例和更高的定量重复性。
二、研究背景:(简要介绍研究进展动态、研究目的和意义)
交联试剂可以将空间距离在一定范围内的化学基团共价连接在一起,定量交联质谱(Quantitative Cross-Linking Mass Spectrometry)可以对交联在一起的氨基酸序列进行鉴定和定量,它是研究蛋白3D结构构象变化和复合体相互作用变化的有力工具。DIA(Data Independent Accquisition)是近年发展起来的非标记蛋白定量方法,与DDA需要依赖质谱一级离子的信号强度信息,只能筛选数十个高丰度离子进行二级碎裂不同,DIA依次将所有一级离子碎裂,并采集全部离子碎裂信息,可利用质谱二级碎裂离子在色谱峰洗脱时间内得到的多个离子信号点形成的峰面积进行定量,具有鉴定更稳定、定量更准确的优点。然而目前对DIA数据进行分析的软件并没有针对交联质谱的数据进行定量分析的常规分析流程,限制了DIA定量技术在定量交联质谱的应用,而DIA技术在定量交联质谱应用的效果如何也未见评估。
三、实验设计:


四、研究成果:(重点图表展示)
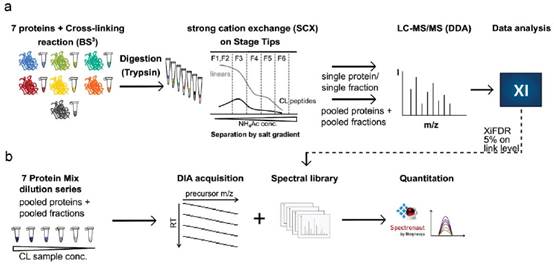
DIA-QCLMS样本制备和数据分析流程:(a)七个蛋白分别用BS3交联,然后分别用胰酶消化,再用强阳离子交换柱进行交联肽段的富集,富集得到的交联肽段用DDA方法做质谱信号采集,数据经Maxquant和Xi分析,并用XiFDR过滤以后用xiDIA-library建立谱图库。(b)七个蛋白的混合肽段梯度稀释成6个浓度,然后一部分直接做DIA检测,另一部分加入到大肠杆菌肽段基质中再做DIA检测。用Spectronaut对DIA数据做定量分析。
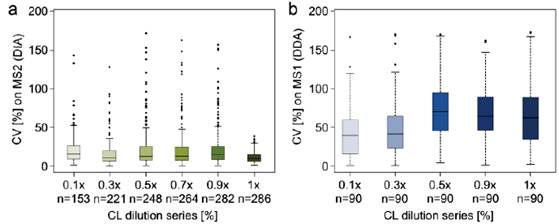
定量交联质谱比较中,有意义的定量值是unique residue pairs (URPs)的信号强度,URP是指交联在一起的氨基酸残基对的唯一匹配质谱信号,它的强度指征蛋白质中交联在一起的位点信号强度。文章主要分析结果如下:
1、比较七个蛋白中鉴定和定量到的URPs,发现DIA方法可以比DDA方法鉴定和定量到更多URPs,且定量值的CV更低。

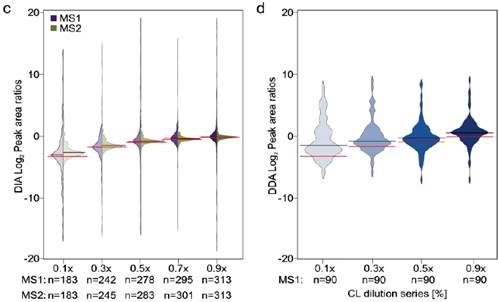
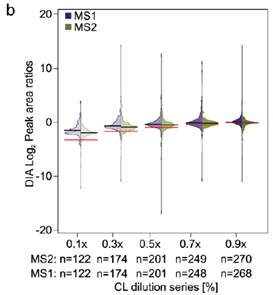
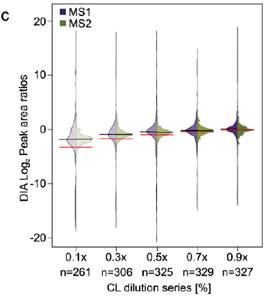
2、比较梯度稀释的七个蛋白混合液用DIA MS2信号定量的CV和用DDA MS1信号定量的CV分布,DIA在不同稀释梯度条件下得到的可定量URPs都比DDA多,CV分布更窄且比DDA低(a,b)。由于是梯度稀释的肽段溶液,比较DIA中分别用MS1,MS2级信号定量得到的值(c黑线)与理论预测值(c红线),以及DDA中MS1级信号定量值(d黑线)与理论预测值(d红线),整体来看,DIA的二级定量信号值最接近理论预测值,即使在浓度稀释到十倍的时候依然与理论预测值很接近,而DDA的定量值与理论存在差距,且随着浓度差异变大,定量的准确性也在降低。
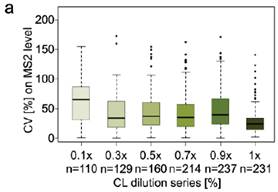
3、当将单独交联得到的七个纯蛋白加入到大肠杆菌的肽段基质中以后再做同样的分析,可以定量的URPs数量相比没有加入基质的时候变少了,而且CV分布也整体变大了(3a与2a比较),理论预测的值与实际定量得到的值的差异也变大了(3b与2c比较),当用Spectronaut中interference correction选项进行校正以后,DIA用MS2,MS1的定量差异几乎不见了,但是理论与实际的定量差异依然存在。表明基质还是还从整体上对DIA-QCLMS定量有一些影响(3c)。
文章亮点(与产品的结合点):
这是一项将DIA技术应用到定量交联质谱的新技术,虽然定量交联质谱可以回答蛋白质组学中,除了整体蛋白丰度以外的蛋白相互作用或蛋白构象信息,可以指征细胞内不同活性的酶,不同调控状态的转录因子,不同状态的相互作用蛋白复合体等等的变化,但是目前这项技术在实际应用中还有许多困难正在克服,比如新的交联试剂的开发、交联数据的解析和假阳性率的控制等。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号