Mol Cell Proteomics. |陈洁| 整合鸟枪法蛋白质组学中鉴定和定量的错误率
大家好,本周分享的是发表在MCP上的一篇关于鸟枪蛋白质组学中的错误率的文章,题目是Integrated identification and quantification error probabilities for shotgun proteomics,作者是瑞典皇家理工学院的Matthew The 和Lukas Kall。
鸟枪法蛋白质组学中蛋白的无标定量存在诸多误差。蛋白的鉴定就是其中的一个来源。然而人们经常忽略某些错误源,且在后续步骤中认为过滤得到的列表是完全正确的,这两个错误的累加很容易导致错误发现率(FDR)失控。另外蛋白定量的错误控制多局限于设置假发现率(FDR)阈值或其他启发式截断(如需要至少一定数量的肽或肽段之间存在一定的相关性)。这并不能直接控制所报告的差异蛋白列表中的错误,还会丢弃仅仅错过其中一个阈值的蛋白,从而丢弃潜在的有价值的信息。而这对于一些蛋白水平的差异表达方法来说就降低了其敏感度。然后通过使用贝叶斯统计,尤其是使用概率图模型,我们就不必再担心这些问题,。
基于以上,作者开发了一种基于概率图模型的软件Triqler(TRansparent Identification-Quantification-Linked Error Rates),它能够整合鉴定和定量的所有步骤中的错误信息,提高定量的敏感度和准确度(Figure 2)。
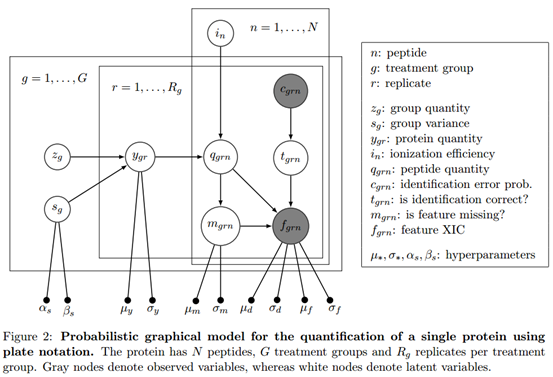
PGMs的一个很好的特性是,由于模型是在参数分布上集成的,所以它们对参数分布的选择通常是鲁棒的(?)。各个参数分布是根据经验分布的形状来选择的。另外,对于缺失值,模型中整合了多个肽段的数据以及蛋白的数量来计算。该模型通过直接计算感兴趣的假设发生的概率来代替各个步骤的分别计算来避免错误的逐级传递带来的FDR不可控的问题。该模型输出不同处理组之间差异倍数变化的后验概率,突出确定性。
作者用了三个数据集进行模型的评估,以说明其特性。三个数据集分中都掺入了已知浓度的蛋白质,分别是(1)iPRG2015研究、(2)iPRG2016研究以及(3)在酵母背景中掺入3种不同浓度的UPS1蛋白质混合物样品。
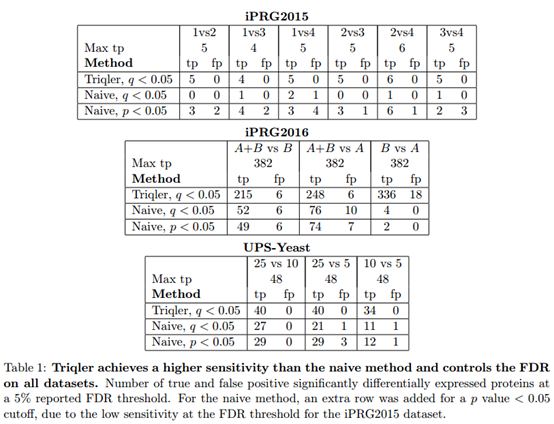
iPRG2015数据集在不同浓度的酵母背景中掺入6种已知的外源蛋白质。iPRG2016数据集包含两个池,即池A和池B的蛋白质片段,称为PrESTs,其中第一个样品仅包含池A PrESTs,第二个样品池包含池B PrESTs,第三个样品池包含两个池的等摩尔混合物。UPS-酵母混合物由3个样品组成,其中UPS1蛋白质混合物分别以25,10和5fmol浓度掺入1μg酵母背景中。 这些数据集中的每一个对于每个样本使用三次重复(Table 1)。
三个数据集均使用Tide进行数据框搜索,并用percolater进行后续处理,然后用percolator的输出作为软件的输入。
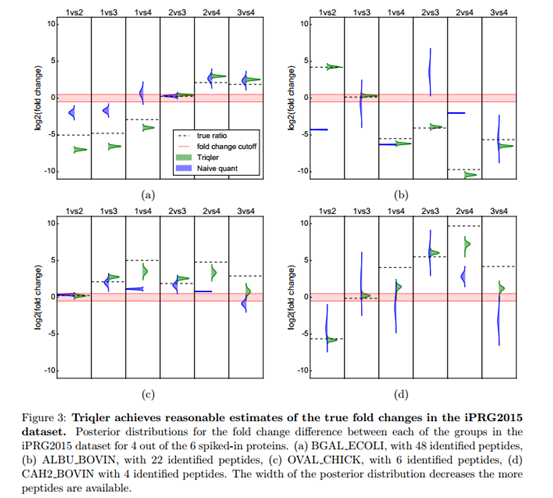
作者分析了3个数据集。在第一个数据集中,用Triqler处理后比用常规方法处理得到的fold change 明显更接近真实值(figure 3)。
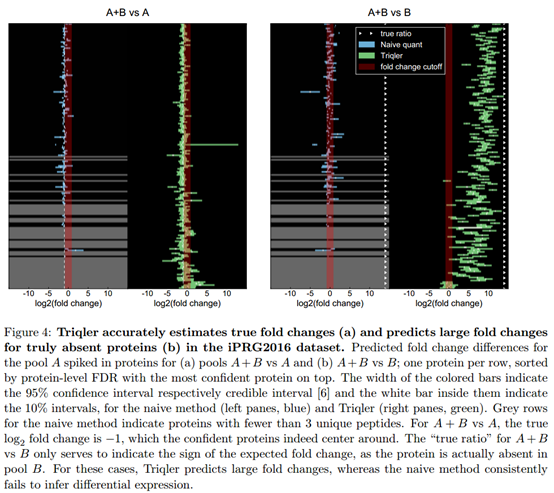
在第二个数据集中,表现了相同的规律,此外,对A+B VS B 中属于A的蛋白的Log(fold change),Triqler显示了显著的差异,而常规方法始终找不到其差异(Figure 4)。
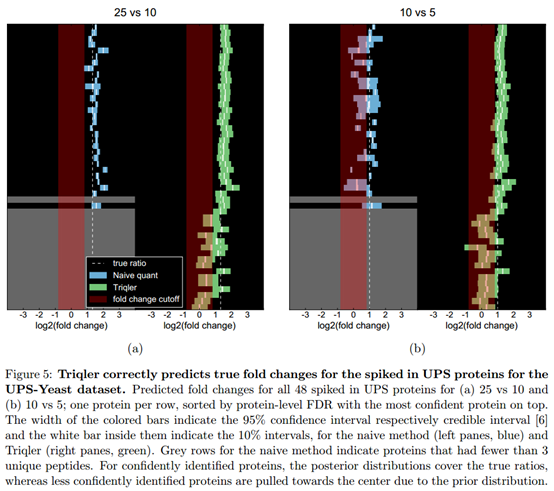
在第三个数据集中,再次观察到随着蛋白质鉴定中的置信度降低,后验分布变宽(Figure 5)。 我们还发现,即使在有许多肽可用的情况下,由于缺失值估算不佳,常规的方法模型仍然会产生假阴性(figure 5b)。
另外,在一个膀胱癌临床数据集(4个患者和4个对照)中,作者在5%的FDR水平发现了35种差异表达蛋白质,而最初的研究在这个阈值上没有发现任何差异蛋白。令人信服的是,这些蛋白在功能注释中富集。
文章特色:使用概率图模型,可以对蛋白更好地定量,有利于寻找不同处理之间差异表达蛋白。另外,对于丰度较低或unique peptides较少的蛋白,这一方法能够起到较大的帮助作用。
文章解读:
文献分享二组-陈洁
文章引用:
doi: 10.1074/mcp.RA118.001018
文章链接:
https://www.mcponline.org/content/18/3/561

 浙公网安备 33010602011771号
浙公网安备 33010602011771号