
文献阅读:me-QTL和p-QTL研究
背景:
GWAS发现的与复杂疾病或性状关联的遗传变异大多数位于非编码区域,而大部分SNP的作用机制尚且未知,所以post-GWAS时代中一个主要的挑战是理解和注释遗传变异与复杂性状/疾病关联的内在机制,而QTL研究是解构这种机制的一种重要研究方法,常见的QTL包括eqtl,pqtl,meqtl,sqtl等,其中最具代表性也是目前应用最广泛的QTL研究范式是表达数量性状座位分析(expression quantitative trait loci, eQTL)。
QTL研究本身具有重要的研究意义,能够阐明DNA序列变异与可定量化的中间分子表型的量化调控关系,同时,这些研究产生的数据资源也同样重要,并被广泛地应用在数量遗传学的方法和应用当中,以解释SNP与各层级分子之间的调控关联以及这种关联对表型的影响机制,例如TWAS,COLCO,MAGMA等方法框架都基于eqtl或pqtl数据开发,并结合疾病GWAS数据来映射疾病相关联的基因,蛋白等,从而揭示疾病发病机制及筛选药物靶点。
目前,关于eqtl的研究相对较多,但关于其他QTL的研究仍然较少或者样本量不足,最近几月,Science和Nature Genetics期刊分别发表了针对大人群样本的pQTL和meQTL的研究,为解释基因组变异到疾病的调控机制和生物过程提供数据支持。
Science:Mapping the proteo-genomic convergence of human diseases
Nature Genetics:Large-scale integration of the plasma proteome with genetics and disease
Nature Genetics:Genetic variation influencing DNA methylation provides insights into molecular mechanisms regulating genomic function
科研启示:
1.一些基于基因的方法学框架完全可以应用在pQTL或meQTL上,例如将基于基因开发的方法框架TWAS应用在蛋白质数据上形成PWAS模型是2021年的一个研究热点,已经或即将发表了数篇关于PWAS的文章。
2.从应用的角度,最容易做的就是整合疾病GWAS与QTL数据进行数据挖掘,可选择的方法学框架主要有MR,PWAS,COLOC等,目前关于疾病GWAS与PQTL数据整合是相对比较热的点,这两篇文章就是模板:文章1,文章2,当然进行这样的运算需要较大的计算资源,如果针对所有蛋白质的GWAS进行MR分析,只下载数据就得6个T。
期刊:
Science (IF=47.73)
Nature Genetics (IF=38.33)
文章1

血浆蛋白质组与遗传学和疾病的大规模整合
研究设计
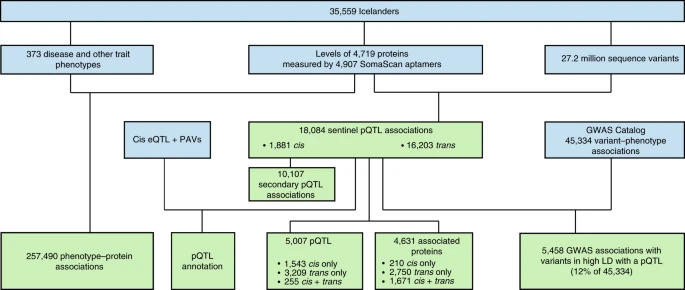
研究结论
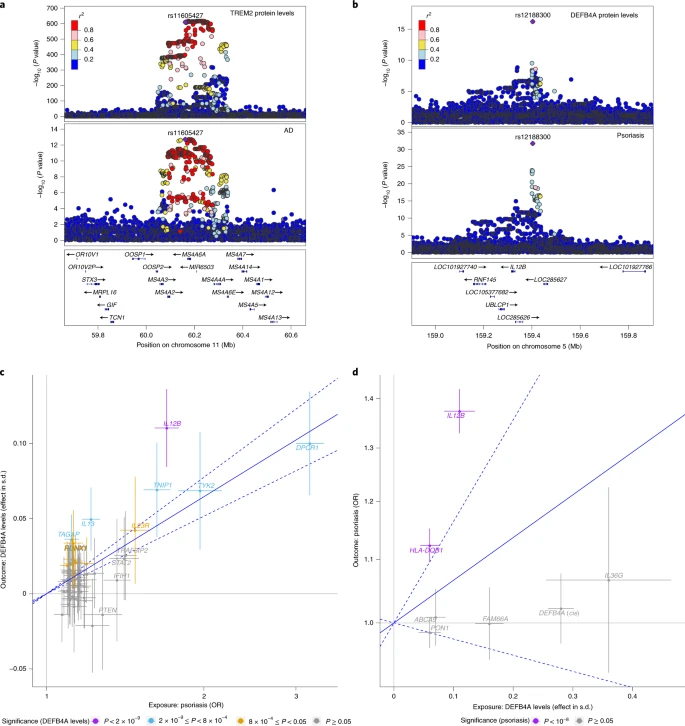
1.蛋白质的全基因组关联分析
- 人群:35,559 冰岛人
- 蛋白测定方法:SomaScan 和 Olink(血浆)
- 4,719 proteins(4,907 aptamers)和2,720 万个变异位点间发现了18,084 个sentinel pQTL关联(P<1.8e-9,conditional test), 其中1,881 个为cis-pQTL,16,203 个为trans-pQTL,分别有 67% 和 96% 是新发现的pqtl.
- 在测量的 4,907 种蛋白质中,4% 仅具有顺式关联,56% 仅具有反式关联,34% 具有顺式和反式关联。只有 6% 没有 pQTL,而 16% 与单个 pQTL 相关,78% 与两个或多个 pQTL 相关
- 数据下载地址:PQTL,数据共6T,每个蛋白的关联分析结果为一个文件。
关联分析方法:we adjusted rank-inverse normal transformed levels for age, sex and sample age for the deCODE Health study on the one hand and the remaining studies on the other hand. We standardized the residuals again using rank-inverse normal transformation and used the standardized values used as phenotypes for genome-wide association testing using the linear mixed model implemented in BOLT-LMM49. We used LD score regression to account for inflation in test statistics due to cryptic relatedness and stratification50. We used a likelihood-ratio test to compute all P values
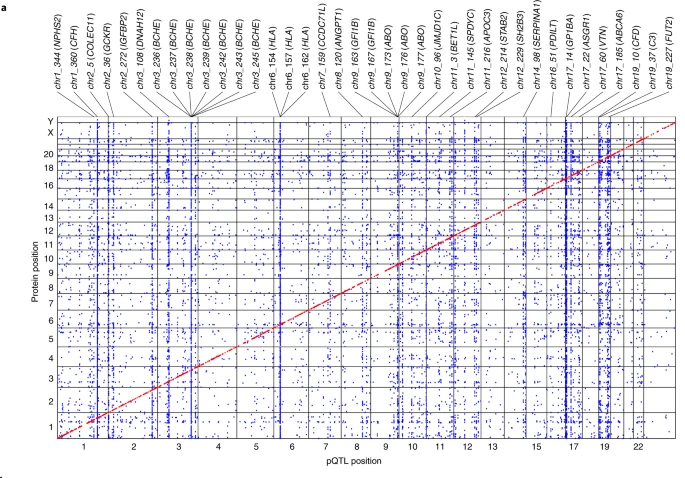
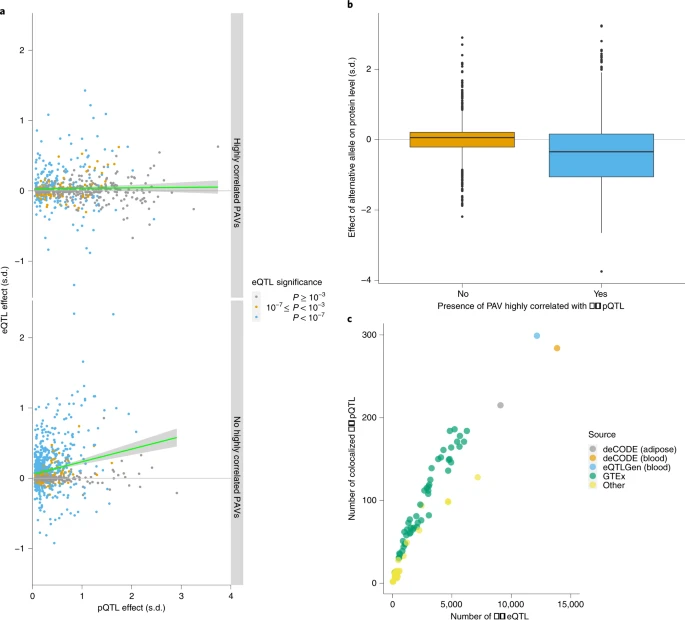
2.pQTL与PAV,eqtl
- 通过将pTQL数据进行注释或与eQTL进行比较,进一步解释pQTL的可能机制
- 在 1,881 个sentinel cis-pQTL 关联中, 27% 仅与 PAV 处于高 LD, 26%仅与cis-eQTL处于高 LD , 8% 与两者均处于高 LD, 632 个(34%)与eQTL具有高 LD 的SNP中,72% 的cis-pQTL和cis-eQTL 具有一致的作用方向(多个组织中eQTL的同一方向>90%),14% 的方向相反(多个组织中eQTL同一方向 <10%),而剩下的 14% 未确定相对方向。
- 在 16,203 个trans-pQTL 中,38 个 trans-pQTL 与 50 种及以上蛋白质相关,这些 pQTL 中的三个(在ABO、GCKR和SH2B3 处)与许多疾病相关。年龄相关性黄斑变性和低密度脂蛋白胆固醇水平分别与这 38 个 pQTL 中的3个和7个相关.
3.血浆pQTL的代表性
此研究是针对血浆样本的,所以作者在文章中也解释了血浆pQTL对组织/细胞特异表达的代表性,发现血浆的 pQTL 对于具有组织特异性表达或致病性的基因仍然具有一定的代表性。
4.pQTL 与 疾病
- 作者提供了一些结合疾病GWAS数据,pQTL数据及其他数据的整合方法,来探究疾病相关蛋白标志物,具体见文章举例。
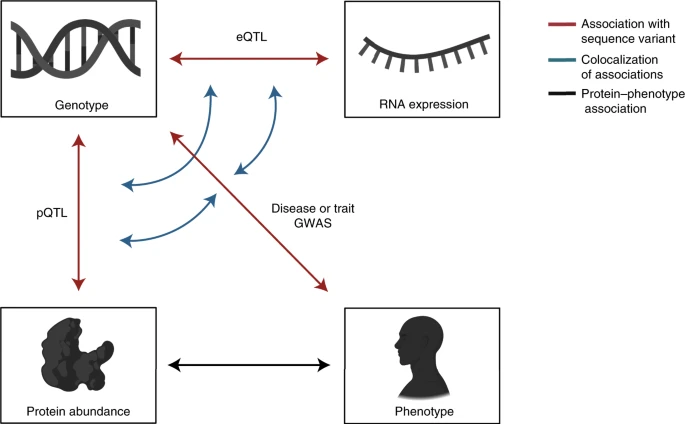
- GWAS Catalog数据库中确定了 45,334 个lead SNP ,其中 5,458 (12%) 个与pQTL处于高 LD(1,223 个cis和 5,125 trans)
- 双向孟德尔随机化分析,可以识别蛋白-疾病的因果关联,可以确定蛋白质水平的改变是疾病的结果还是其发病机制的一部分。双向孟德尔随机化分析表明 DEFB4A 水平升高是银屑病的结果,DEFB4A 不直接参与其发病机制
####################################################################################
文章2:

影响DNA甲基化的遗传变异为调节基因组功能的分子机制提供见解
核心研究机构:德国环境健康研究中心计算生物研究所与英国伦敦帝国理工学院流行病学与生物统计学系
研究摘要
该研究报道了迄今最大规模的多种群、多细胞类型的人类全血样本DNA甲基化QTL(meQTL)图谱,并对DNA甲基化调控元件进行了深入分析,同时还将meQTL与其他分子表型QTL研究及疾病表型GWAS研究进行了整合,系统性地揭示了由DNA甲基化所介导的人类遗传变异与各类分子或疾病表型之间的调控机制,从而为参与调控的分子网络以及将遗传变异与人类表型联系起来的潜在途径提供了新的见解。
研究设计
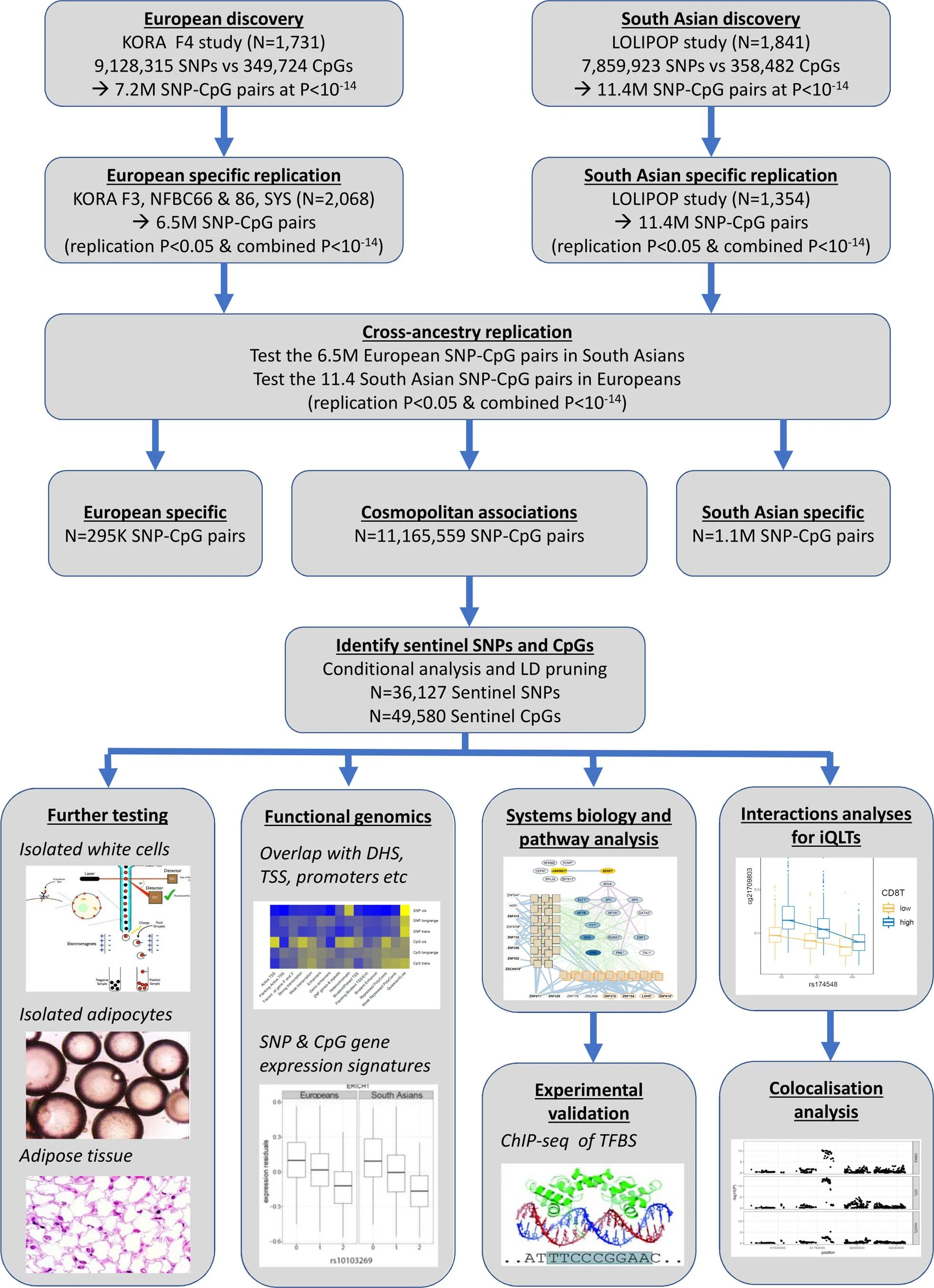
结论
1.DNA甲基化的全基因组关联分析
- 欧洲人:3799(发现队列:1731;验证队列:2068);南亚人:3195(发现队列:1841,验证队列:1354)
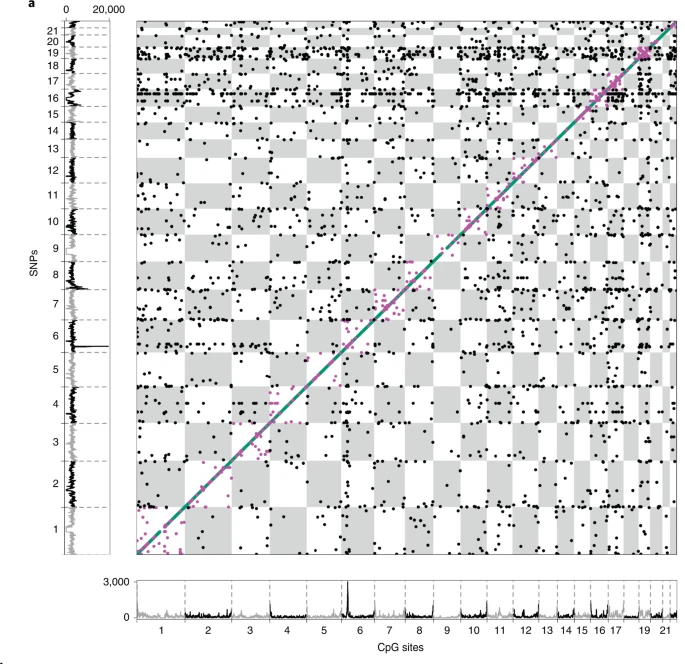
- 全血样本,产生了一个包含 2,709,428 个 SNP 和 70,709 个 CpG 位点组成的11,165,559 个 meQTL pairs数据集(下载地址:https://zenodo.org/record/5196216#.YdQFzNpBxEZ)。
- 其中:
cis-meQTL:10,346,172 pairs(2,650,691 SNPs and 67,694 CpG sites)
long-range cis-meQTL:351,472 pairs(120,593 SNPs and 1,846 CpG sites)
trans-meQTL:467,915 pairs(200,761 SNPs and 3,592 CpG sites). - 平均而言,SNP 解释了 CpG 位点甲基化变异的 10.3%(四分位距,4.4-11.5%)
关联分析中,进行独立队列内部数据归一化,以协变量拟合每个CpG位点的甲基化百分比,然后用基因型拟合残差并执行最后的检验,CpG ~ Covariates,CpGresiduals ~ SNPgenotype
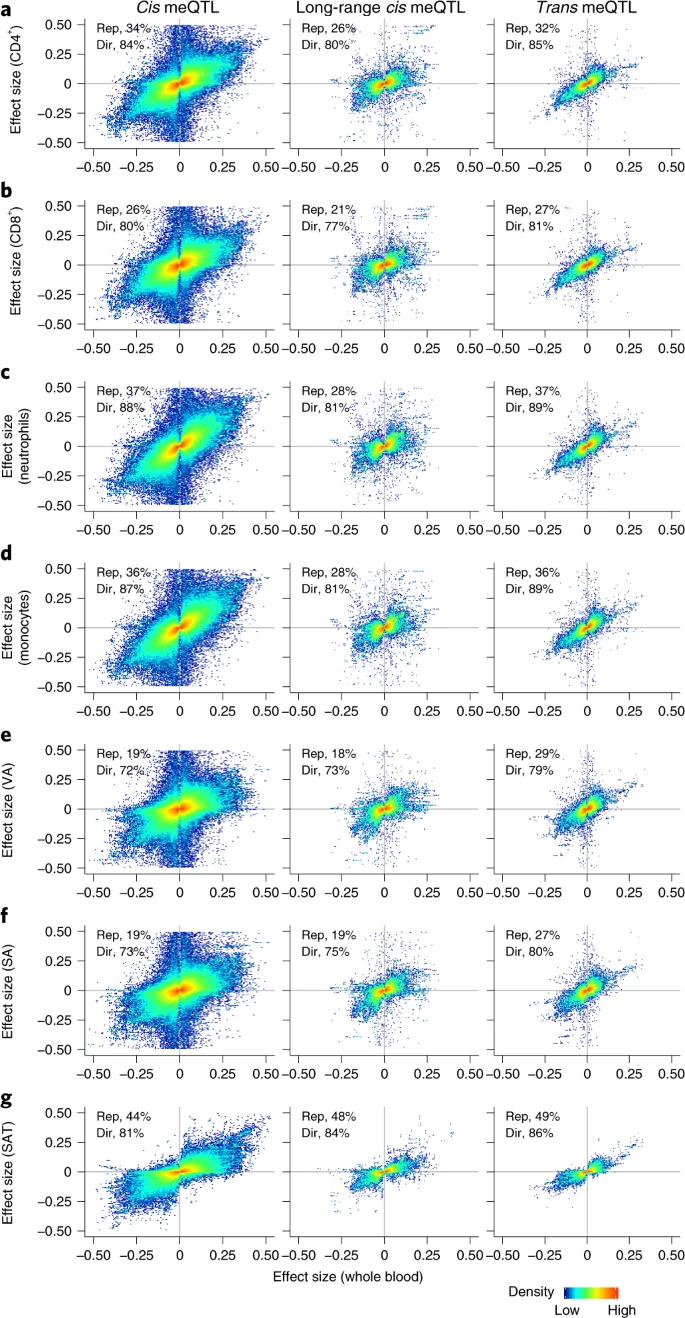
2.meQTL 在不同的细胞类型
上面的meQTL是基于全血,作者进一步通过分离的细胞进行验证,以检验meqtl在不同细胞类型中的分布。验证标准:P < 0.05 且效应方向相同,发现许多meQTL在不同的细胞谱系中发挥作用,可能与血液以外的组织和生物系统相关。
- 分离的白细胞亚群(n=60)验证了26-37%
- 分离的内脏脂肪细胞(n=48)验证了19.2%
- 分离的皮下脂肪细胞(n=48)验证了19.4%
- 脂肪组织(n=603)44.2%
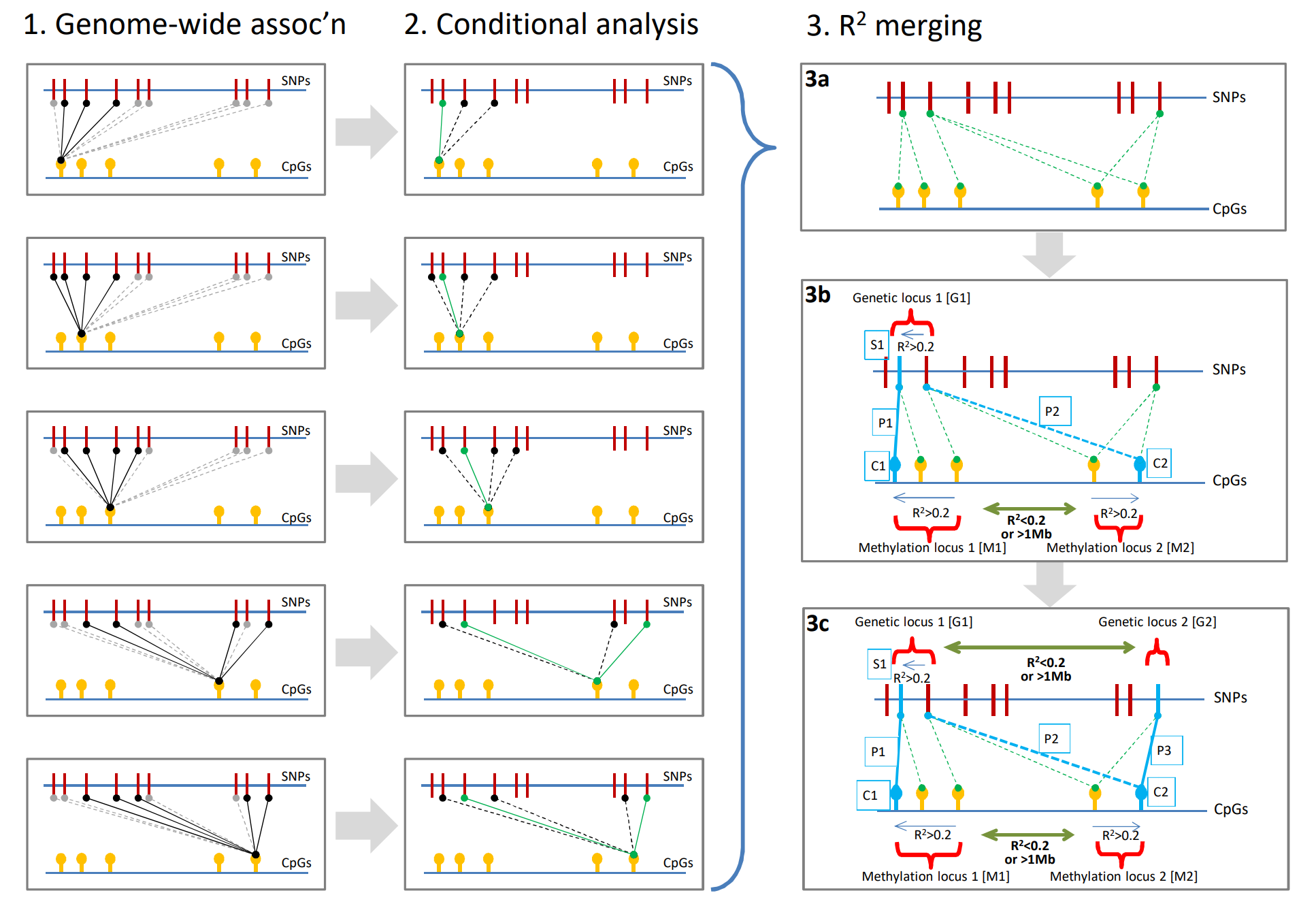
3.独立cis-meQTL的识别
- 方法:Conditional test 和 Locus combination
cis-meQTL: 34,001 independent genetic loci ~ 46,664 independent methylation loci
long-range cis-meQTL: 467 independent genetic loci ~ 499 independent methylation loci
trans-meQTL: 1,847 independent genetic loci ~ 3,020 independent methylation loci.
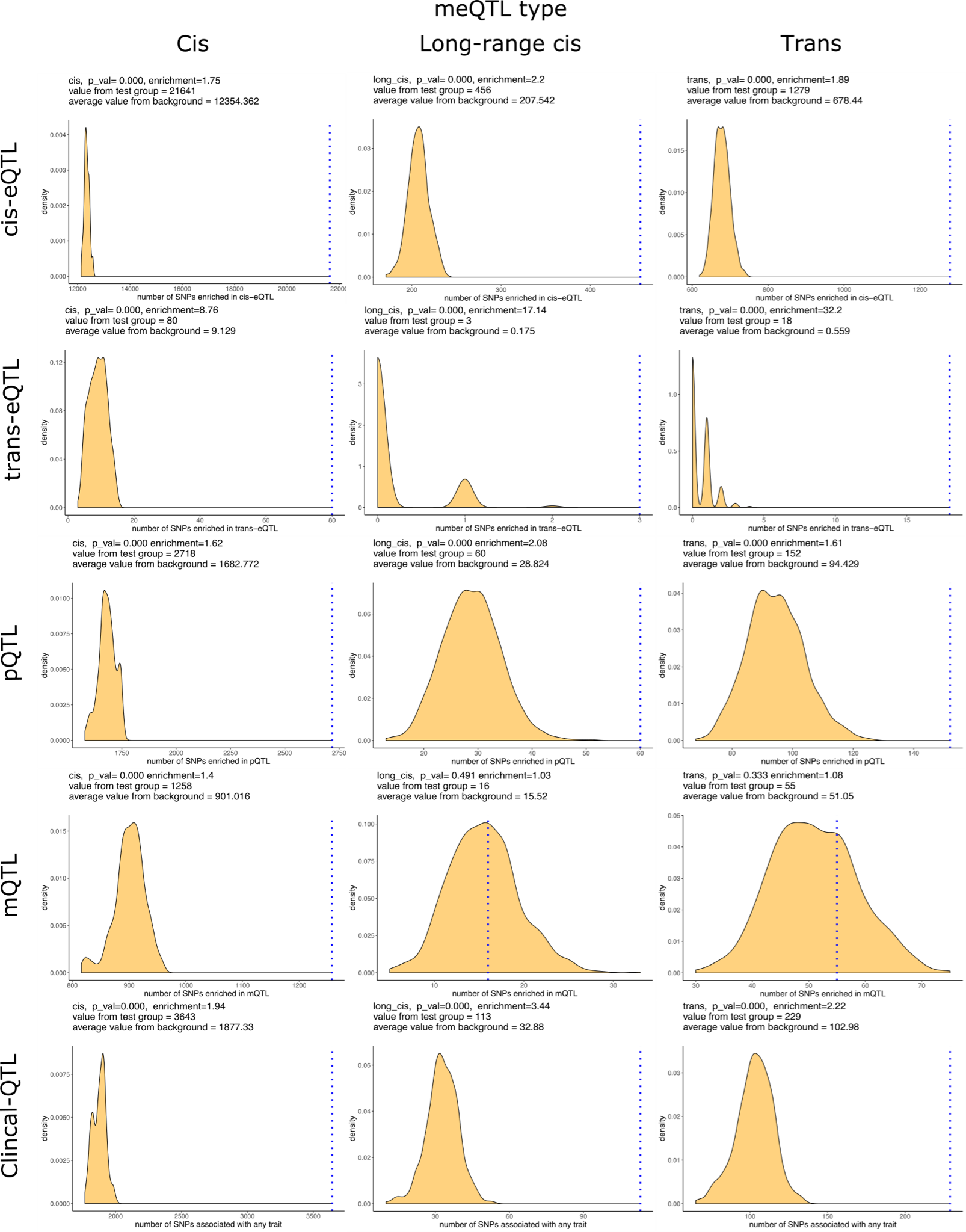
4.meQTL 与其他分子或表型的关联
- 富集分析:排列检验分析meQTL在不同染色质状态,eQTL,pQTL,mQTL,phenoQTL中的富集,形成 meQTL 对的 SNP 和 CpG 位点都富含多种功能相关特征,包括共享染色质状态、Hi-C 相互作用、顺式或反式基因表达以及与多种代谢和临床特征。
- 分子关联:作者也探讨了反式表达数量性状甲基化位点,eQTM,研究 DNA 甲基化和基因表达之间的反式作用关系。
- 因果推断:使用的 meQTL 作为遗传工具来检查 DNA 甲基化和体重指数 (BMI) 之间的潜在因果关系,发现 meQTL SNP 和 CpG 位点都强烈富集于与 BMI 的关联的位点,SMR 表明 DNA 甲基化和 BMI 在 374 个位点之间存在潜在的因果关系,其中 239 个显示了共享因果变体的证据(coloc PP4 > 0.6)。
5.研究举例
- 作者选择部分基因作为更为细致的解释和说明:NFKBIE,MGA,COMMD7,SENP7,ZNF333
- 在最后,作者进一步补充了 meQTL 的人群特异性以及meQTL与环境的相互作用分析

