De novo molecular design and generative models
1.摘要
分子设计策略是药物发现的组成部分。在过去的三十年里,分子从头设计的计算方法已经被开发出来。最近,随着机器学习(ML)和人工智能(AI)的进步,药物发现领域获得了新的实践经验。在这里,我们回顾了这些经验,并根据分子表示的程度,介绍了从头设计的方法:即基于原子、基于片段、基于反应的分子设计方法(atom-based, fragment-based, or reaction-based paradigm)。此外,我们强调了评估基准的价值(value of strong benchmarks),描述了在实践中使用这些方法的主要挑战,并对未来几年进一步探索的机会和需要解决的挑战提出了我们的观点。
2.前言
分子从头设计是自动提出新的化学结构、以最佳方式满足所需的分子特征的过程。通常在药物发现中,目标分子特征是为了获得理想的生物学效应(biological response),同时保持可接受的药代动力学特性(pharmacokinetic properties)。最近,从头设计也被称为生成化学,这源于人工智能中生成模型的日益普及。
传统上,虚拟筛选(VS)是用来识别可能表现出理想的实验结果的分子。与从头设计相比,一个关键的区别是所考虑的分子来源:在虚拟筛选中,结构是预先知道的;而在从头设计中,我们试图生成待评估的结构。
化学空间(即横跨所有可能的分子的广阔空间)是巨大的。尽管按照药物发现的标准,虚拟筛选库已经变得非常庞大(许多库中含有多达10亿个分子),但这些库所对应的化学空间只占很小一部分。当考虑这样的化合物库时,评估方法可能会必然牺牲预测的准确性。通过使用从头设计以定向方式生成化合物,计算工作者希望更有效地穿越化学空间,在分析比大型化学库(“粗暴”筛选)更少的分子的同时,获得最佳的化学解决方案(图1)。此外,对于一个给定的目标,化学空间可能有许多可接受的区域,为此,分子设计方法的任务是平衡对全局解决方案的探索和对局部最小值的利用。
从头设计在化学信息学中有着丰富的历史。随着ML方法不断为大型搜索空间的导航和取样提供新的可能性,从头设计最近也受到了关注。在这篇文章中,我们从分子表征程度(coarseness粗糙性)的角度来考虑从头设计的方法。具体而言,我们对生成新结构的基于原子的、基于片段的和基于反应的方法进行了区分。在讨论生成化学已建立的方法和新的前沿领域之前,我们首先回顾了评估比较能力(比较从头设计方法的能力)的方法。最后,我们评估了从头设计的成功之处,并强调了在实现分子从头设计全部潜力的道路上有待跨越的潜在障碍。
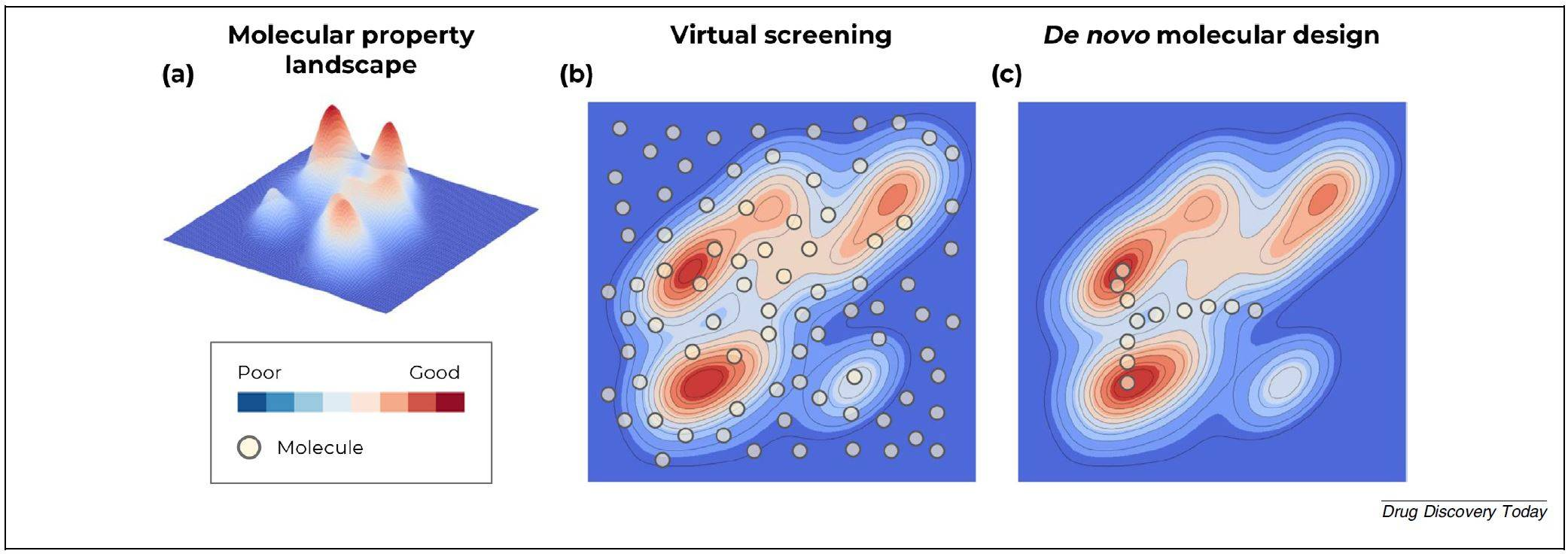
(a)在给定一个任意的客观分子特性轮廓的情况下,用颜色表示该区域内分子的最优性。对预先存在的大型化学库进行虚拟筛选(b)和通过有效的从头分子设计程序穿越化学空间(c)之间的概念差异。从头设计的结果是考虑更少的分子,通过更有效地遍历化学空间达到最佳的分子结构。
3.分子设计
3.1评估从头分子设计的方法
为了一致地评估自动生成化学结构的方法的进展,建立评估标准的基准是至关重要的。
从头设计方法通常是通过其在独立的任务上的表现来评估的,如进行最大程度类药性的定量(QED)或计算辛醇-水分配系数(ClogP)。尽管这对于展示优化器生成分子的能力来说是微不足道的,但这样的基准未能捕捉到现实世界药物发现的复杂性。
相比之下,评估从头设计方法的另一种方法是通过实验来证明它们的使用。例如,Firth等人合成并测试了循环依赖性激酶2(CDK2)的新型抑制剂,虽然作者追求前瞻性验证的做法值得赞扬,但对该方法的评估是有问题的,因为结果是传闻,而且取决于与从头设计算法无关的许多因素。
3.2分子表示
评估化学结构的计算方法必须依赖于合适的分子表示,也就是后续算法所看到的分子结构的形式。分子表示是一个广泛的话题。例如,方法可以编码官能团的存在或不存在,将分子表达为其拓扑图,或包括描述键角的三维信息。
在从头设计的方法中,常见的分子表征是基于文本的,如SMILES,以及基于图形的(其中分子生成器可能明确地操作分子拓扑结构)。基于文本的方法得益于自然语言处理(NLP)的大量活跃研究,而基于图形的方法则体现了对分子结构更自然的表述。其他影响表示法选择的因素包括分子表示法是否是离散的(如比特向量)、连续的(如浮点向量)和可逆的。最近对从头设计方法的评论集中在通过生成模型结构的角度来讨论分子表示法,而我们在这里集中讨论分子表示法的颗粒度(图2),因为这直接转化为分子设计的实际方面。
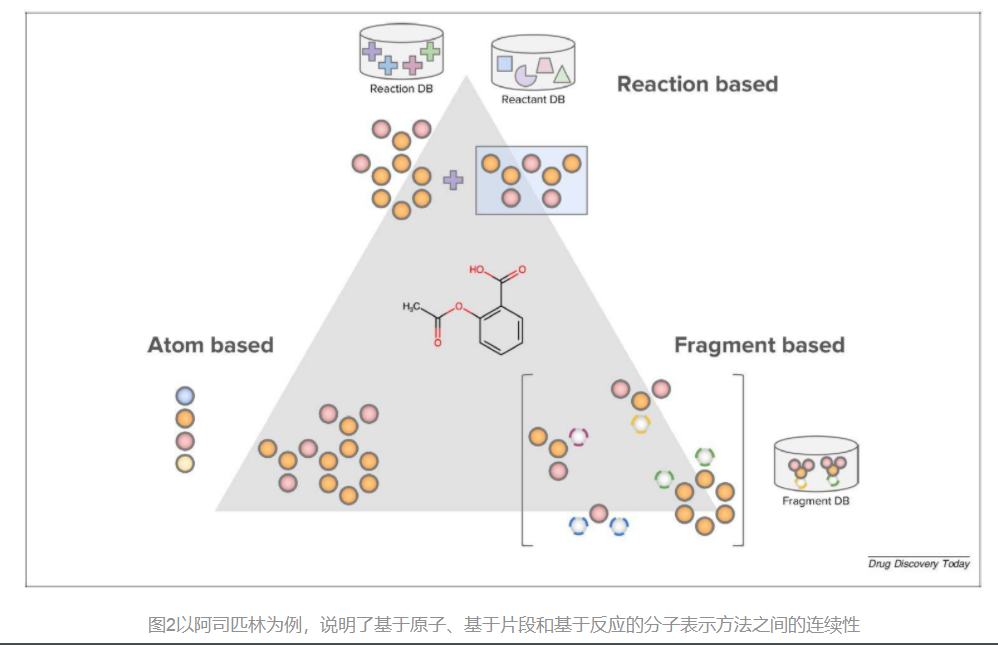
分子表示方法:基于原子的方法是由一个包含少量原子和键的“词汇”支持的。基于反应的方法是由dual sets of reactants和反应规则支持的。最后,基于片段的方法由一个片段方案和一组可互换的片段支持;灰色的原子表示用断开类型(颜色)注释的连接点。
SMILES作为生成模型的分子表示法已经很普遍了;然而,SMILES的一个缺点是每个SMILES字符串并不是对分子图的唯一描述。一个SMILES是通过分子结构的线性行走来构建的;因此,不同的起始位置和通过分子的路径会产生不同的SMILES。经典的SMILES(Canonical SMILES)代表了分子图的标准化遍历;然而,在经典的SMILES上训练的生成模型可以捕获SMILES语法的干扰方面,而不是基础的分子结构。研究表明,在生成模型的训练中纳入非经典的SMILES是有好处的。此外,从适应性来说,SMILES已经被描述为更适合与ML一起使用,包括DeepSMILES和SELFIES。
(总结:一个分子的SMILES不唯一,canonical SMILES是唯一的,但是基于它学到的模型捕获的只是SMILES的令人讨厌的东西。所以还是普通的SMILES更适合一些。一些新的改进也不错,例如deepsmiles和selfies。)
化学结构可以在原子水平上表示(通过对分子中的每个原子和键进行编码),或者更粗略的表示(功能团或子结构及其连接性保持固定,如具有1,3取代模式的苯基可以被视为一个单一的基团)。进一步的延展,是对反应进行编码,即目标分子被认为是反应物和反应条件的产物。在实践中,基于原子、基于片段和基于反应的方法都有明显的优势和劣势,许多方法模糊了这些分类之间的界限。
4.无梯度分子优化
给定分子表示,优化算法根据可计算的目标函数指导生成最优分子。用于从头设计的元启发式("无梯度")Metaheuristic ('gradient-free')方法使用基于种群population-based的随机优化程序来导航化学空间,如进化算法或群体智能swarm intelligence。简而言之,我们突出了最近文献中关于其选择的分子表示粒度的示范性工作。(见表1)。

4.1基于原子
基于原子的从头设计方法的一个例子是基于图的遗传算法(GB-GA)graph-based genetic algorithm,它使用反应SMARTS对候选分子库进行编译和交叉,同时自然选择程序确保最优化的分子保持在群体中;ChemGE使用语法进化grammatical evolution来优化一个符合上下文无关语法的SMILES种群。GBGA被纳入了GuacaMol基准,并取得了最先进的性能,尽管基准的作者通过测量分子中活性和不稳定基团的数量来评价化合物的“质量”。
Winter等描述了分子群优化(MSO),一种利用粒子群优化来在连续嵌入空间中【识别理想区域】,然后解码离散分子结构的方法。虽然MSO使用的表示是学习的,但优化过程是一个无梯度的方法,并在GuacaMol目标导向基准上实现了最先进的性能。以群体为基础的方法的一个关键问题是保持群体的结构多样性。MolFinder使用池中分子之间的最小拓扑距离来确保这一点,而基于图的GB-EPI扩展了GB-GA,来维持基于特征的小生态位的种群。
4.2基于片段
基于片段的方法限制了新化合物的生成。其包括已知的相关子结构,如药物化学文献中的子结构。片段化方案使用简单的规则(如所有的无环单键)或受逆合成启发的断裂retrosynthetically inspired disconnections来解构分子,然后可以使用每个包含一个或多个原子的片段库来构建新分子。
MOARF是一种基于片段的从头设计方法,它利用了一套逆向合成断开规则(SynDiR)和一个进化算法。最近开发的CReM框架使用化学上合理的突变,使用从匹配的分子对中改编的片段方案,在GuacaMol目标导向的基准任务上显示出与MSO相当的性能。
4.3基于反应
可以说,从头设计的最实用的策略是在计算机中进行正向反应。2003年,Vinkers等人描述了SYNOPSIS,这种方法迭代地使用虚拟反应来最大化期望的【适应度函数】,通过合成和测试旨在抑制艾滋病毒逆转录酶(HIV-RT)的化合物来证明。最近开发的AutoGrow4利用了一种遗传算法和一个反应库,该反应库来自于稳健的有机反应,用来突变种群中的分子。
反应模板的一个缺点是不考虑分子中其他反应基团,而匹配反应处理,这在实际中会影响反应。Ghiandoni等人最近报告了一个反应类别推荐器,允许从不需要的类别中过滤反应。
5.基于梯度的分子优化
尽管基于群体的元启发式设计方法在寻找优化最小值方面已被证明是稳健的,但在过去的三年里,分子设计的深度学习方法已被广泛采用。基于梯度的ML方法通常在现有分子结构的大型语料库中进行预训练,并学习如何在任意的属性表面导航,以获得最佳解决方案。
研究人员已经提出了几种用于学习生成分子结构的深度学习架构,包括变异自动编码器(VAEs)、生成对抗网络(GANs)和循环神经网络(RNNs)。一旦经过训练,生成模型允许用户从所学的化学空间中抽取分子,当与优化过程(如贝叶斯优化(BO)或强化学习(RL))相结合时,可以有效地识别理想的分子轮廓。关于所涉及的ML过程的更多细节,我们请读者参考最近的文章(参考文献9-11)。
5.1基于原子
许多基于原子的生成模型利用SMILES作为分子表示。鉴于SMILES是一种基于文本的表示方法,生成化学方法能够利用适合序列的深度学习架构,如RNNs。通过对大量的分子结构语料库进行预训练,生成模型可以先验地学习,从而囊括有效的SMILES的语法和句法。早期的工作是用转移学习来使生成偏向于感兴趣的化学空间,现在普遍的做法是将生成任务与RL算法结合起来,后者学习在搜索空间中导航以获得更高的奖励(更多的最佳分子也有可能是更好的分子)。
除了基于SMILES的生成模型外,人们对直接考虑分子图的拓扑结构的模型也很感兴趣,其中原子和键分别被视为节点和边。通过对分子结构更自然的表述,基于图的模型试图避开SMILES语法的人为方面的因素。GraphVAE和MolGAN是基于生成图的方法,它可以一次学会生成整个图的邻接矩阵。还有人描述了通过迭代修改分子图来逐步学习生成分子的方法。最近,RL方法在图的设置中显示了有希望的结果。
5.2基于片段
虽然经过预训练的基于原子的生成模型对其训练数据中存在的子结构保持了很强的先验能力,但它们仍然能够单独修改分子中的每个原子。尽管这种灵活性鼓励学习的模型具有最大的表现力,从而对化学空间有更广泛的覆盖,但基于片段的方法使用更粗糙的分子表示法来限制搜索空间(1996年文献)。
20多年后,葛兰素史克公司的研究人员报告了一个使用Seq2Seq模型的情况,该模型可以学习到还原图和SMILES之间的转换。Jin等人描述了JTVAE,这是一个两步的生成过程,首先构建一个结点树来表示分子中的分子子结构的组成(很像还原图),随后,使用图信息传递网络来解码最终的分子结构。DeepFMPO通过所考虑的片段的相似性来限制优化,显示出更好的性能。
5.3基于反应
有几项工作报告了用于新分子设计的基于反应的生成模型。DINGOS以美国专利商标局(USPTO)的反应数据集为基础,使用ML和基于规则的混合方法产生与分子模板结构相似的新化学实体。Molecule Chef使用VAE嵌入反应物结构,并通过偏重反应物的选择(对于单步反应)来优化生成物的分子特性。
ChemBO代表了一种算法上更简单的方法,即用随机选择的反应物和条件来随机生成候选结构,然后对其进行性能评估。这个工作流程通过多步化学合成产生分子,尽管反应物的选择不偏向于优化目标。
最近的研究报告称,通过将正向合成建模为马尔科夫决策过程(MDP),使用RL来浏览可能反应的巨大空间。REACTOR使用一组双反应物的反应模板(编码为反应SMARTS),并根据哪些反应物能最大限度地提高下一个状态的获益来选择缺失的反应物。当产生一个以上的反应产物时,会选择与最大获益相关的产物。同样,前向合成的政策梯度(PGFS)将有偏见的反应物选择与多步反应相结合,(分子)图(DoGs)的有向无环图(DAG)方法迭代地生成前向合成路线的DAG。
在药物发现方面,ML的突破性成功之一是合成路线规划的发展,它可以学习从大型反应数据库中预测合成路线。目前基于反应的生成模型的一个限制是它们对“手工”制作的反应模板的依赖。我们希望未来的研究能将学习到的反应模式与化学空间的通用优化器结合起来。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号