R语言学习 - 热图美化
实际应用中,异常值的出现会毁掉一张热图。这通常不是我们想要的。为了更好的可视化效果,需要对数据做些预处理,主要有对数转换,Z-score转换,抹去异常值,非线性颜色等方式。
对数转换
为了方便描述,假设下面的数据是基因表达数据,4个基因 (a, b, c, d)和5个样品 (Grp_1, Grp_2, Grp_3, Grp_4),矩阵中的值代表基因表达FPKM值。
data <- c(rnorm(5,mean=5), rnorm(5,mean=20), rnorm(5, mean=100), c(600,700,800,900,10000))
data <- matrix(data, ncol=5, byrow=T)
data <- as.data.frame(data)
rownames(data) <- letters[1:4]
colnames(data) <- paste("Grp", 1:5, sep="_")
data
Grp_1 Grp_2 Grp_3 Grp_4 Grp_5
a 6.61047 20.946720 100.133106 600.000000 5.267921
b 20.80792 99.865962 700.000000 3.737228 19.289715
c 100.06930 800.000000 6.252753 21.464081 98.607518
d 900.00000 3.362886 20.334078 101.117728 10000.000000
# 对数转换
# +1是为了防止对0取对数;是加1还是加个更小的值取决于数据的分布。
# 加的值一般认为是检测的低阈值,低于这个值的数字之间的差异可以忽略。
data_log <- log2(data+1)
data_log
Grp_1 Grp_2 Grp_3 Grp_4 Grp_5
a 2.927986 4.455933 6.660112 9.231221 2.647987
b 4.446780 6.656296 9.453271 2.244043 4.342677
c 6.659201 9.645658 2.858529 4.489548 6.638183
d 9.815383 2.125283 4.415088 6.674090 13.287857
data_log$ID = rownames(data_log)
data_log_m = melt(data_log, id.vars=c("ID"))
p <- ggplot(data_log_m, aes(x=variable,y=ID)) + xlab("samples") + ylab(NULL) + theme_bw() + theme(panel.grid.major = element_blank()) + theme(legend.key=element_blank()) + theme(axis.text.x=element_text(angle=45,hjust=1, vjust=1)) + theme(legend.position="top") + geom_tile(aes(fill=value)) + scale_fill_gradient(low = "white", high = "red")
ggsave(p, filename="heatmap_log.pdf", width=8, height=12, units=c("cm"),colormodel="srgb")
对数转换后的数据,看起来就清晰的多了。而且对数转换后,数据还保留着之前的变化趋势,不只是基因在不同样品之间的表达可比 (同一行的不同列),不同基因在同一样品的值也可比 (同一列的不同行) (不同基因之间比较表达值存在理论上的问题,即便是按照长度标准化之后的FPKM也不代表基因之间是完全可比的)。
Z-score转换
Z-score又称为标准分数,是一组数中的每个数减去这一组数的平均值再除以这一组数的标准差,代表的是原始分数距离原始平均值的距离,以标准差为单位。可以对不同分布的各原始分数进行比较,用来反映数据的相对变化趋势,而非绝对变化量。
data_ori <- "Grp_1;Grp_2;Grp_3;Grp_4;Grp_5
a;6.6;20.9;100.1;600.0;5.2
b;20.8;99.8;700.0;3.7;19.2
c;100.0;800.0;6.2;21.4;98.6
d;900;3.3;20.3;101.1;10000"
data <- read.table(text=data_ori, header=T, row.names=1, sep=";", quote="")
# 去掉方差为0的行,也就是值全都一致的行
data <- data[apply(data,1,var)!=0,]
data
Grp_1 Grp_2 Grp_3 Grp_4 Grp_5
a 6.6 20.9 100.1 600.0 5.2
b 20.8 99.8 700.0 3.7 19.2
c 100.0 800.0 6.2 21.4 98.6
d 900.0 3.3 20.3 101.1 10000.0
# 标准化数据,获得Z-score,并转换为data.frame
data_scale <- as.data.frame(t(apply(data,1,scale)))
# 重命名列
colnames(data_scale) <- colnames(data)
data_scale
Grp_1 Grp_2 Grp_3 Grp_4 Grp_5
a -0.5456953 -0.4899405 -0.1811446 1.7679341 -0.5511538
b -0.4940465 -0.2301542 1.7747592 -0.5511674 -0.4993911
c -0.3139042 1.7740182 -0.5936858 -0.5483481 -0.3180801
d -0.2983707 -0.5033986 -0.4995116 -0.4810369 1.7823177
data_scale$ID = rownames(data_scale)
data_scale_m = melt(data_scale, id.vars=c("ID"))
p <- ggplot(data_scale_m, aes(x=variable,y=ID)) + xlab("samples") + ylab(NULL) + theme_bw() + theme(panel.grid.major = element_blank()) + theme(legend.key=element_blank()) + theme(axis.text.x=element_text(angle=45,hjust=1, vjust=1)) + geom_tile(aes(fill=value)) + scale_fill_gradient(low = "white", high = "red")
ggsave(p, filename="heatmap_scale.pdf", width=8, height=12, units=c("cm"),colormodel="srgb")
Z-score转换后,颜色分布也相对均一了,每个基因在不同样品之间的表达的高低一目了然。但是不同基因之间就完全不可比了。

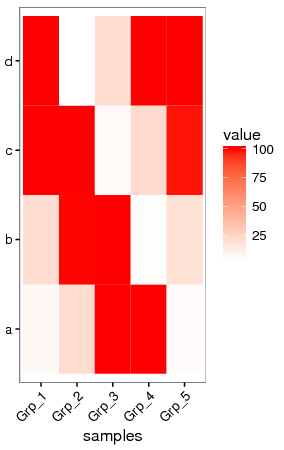
抹去异常值
粗暴一点,假设检测饱和度为100,大于100的值都视为100对待。
data_ori <- "Grp_1;Grp_2;Grp_3;Grp_4;Grp_5
a;6.6;20.9;100.1;600.0;5.2
b;20.8;99.8;700.0;3.7;19.2
c;100.0;800.0;6.2;21.4;98.6
d;900;3.3;20.3;101.1;10000"
data <- read.table(text=data_ori, header=T, row.names=1, sep=";", quote="")
data[data>100] <- 100
data
Grp_1 Grp_2 Grp_3 Grp_4 Grp_5
a 6.6 20.9 100.0 100.0 5.2
b 20.8 99.8 100.0 3.7 19.2
c 100.0 100.0 6.2 21.4 98.6
d 100.0 3.3 20.3 100.0 100.0
data$ID = rownames(data)
data_m = melt(data, id.vars=c("ID"))
p <- ggplot(data_m, aes(x=variable,y=ID)) + xlab("samples") + ylab(NULL) + theme_bw() + theme(panel.grid.major = element_blank()) + theme(legend.key=element_blank()) + theme(axis.text.x=element_text(angle=45,hjust=1, vjust=1)) + geom_tile(aes(fill=value)) + scale_fill_gradient(low = "white", high = "red")
ggsave(p, filename="heatmap_nooutlier.pdf", width=8, height=12, units=c("cm"),colormodel="srgb")
虽然损失了一部分信息,但整体模式还是出来了。只是在选择异常值标准时需要根据实际确认。
非线性颜色
正常来讲,颜色的赋予在最小值到最大值之间是均匀分布的。非线性颜色则是对数据比较小但密集的地方赋予更多颜色,数据大但分布散的地方赋予更少颜色,这样既能加大区分度,又最小的影响原始数值。通常可以根据数据模式,手动设置颜色区间。为了方便自动化处理,我一般选择用四分位数的方式设置颜色区间。
data_ori <- "Grp_1;Grp_2;Grp_3;Grp_4;Grp_5
a;6.6;20.9;100.1;600.0;5.2
b;20.8;99.8;700.0;3.7;19.2
c;100.0;800.0;6.2;21.4;98.6
d;900;3.3;20.3;101.1;10000"
data <- read.table(text=data_ori, header=T, row.names=1, sep=";", quote="")
data$ID = rownames(data)
data_m = melt(data, id.vars=c("ID"))
# 获取数据的最大、最小、第一四分位数、中位数、第三四分位数
summary_v <- summary(data_m$value)
summary_v
Min. 1st Qu. Median Mean 3rd Qu. Max.
3.30 16.05 60.00 681.40 225.80 10000.00
# 在最小值和第一四分位数之间划出6个区间,第一四分位数和中位数之间划出6个区间,中位数和第三四分位数之间划出5个区间,最后的数划出5个区间
break_v <- unique(c(seq(summary_v[1]*0.95,summary_v[2],length=6),seq(summary_v[2],summary_v[3],length=6),seq(summary_v[3],summary_v[5],length=5),seq(summary_v[5],summary_v[6]*1.05,length=5)))
break_v
[1] 3.135 5.718 8.301 10.884 13.467 16.050 24.840
[8] 33.630 42.420 51.210 60.000 101.450 142.900 184.350
[15] 225.800 2794.350 5362.900 7931.450 10500.000
# 安照设定的区间分割数据
# 原始数据替换为了其所在的区间的数值
data_m$value <- cut(data_m$value, breaks=break_v,labels=break_v[2:length(break_v)])
break_v=unique(data_m$value)
data_m
ID variable value
1 a Grp_1 8.301
2 b Grp_1 24.84
3 c Grp_1 101.45
4 d Grp_1 2794.35
5 a Grp_2 24.84
6 b Grp_2 101.45
7 c Grp_2 2794.35
8 d Grp_2 5.718
9 a Grp_3 101.45
10 b Grp_3 2794.35
11 c Grp_3 8.301
12 d Grp_3 24.84
13 a Grp_4 2794.35
14 b Grp_4 5.718
15 c Grp_4 24.84
16 d Grp_4 101.45
17 a Grp_5 5.718
18 b Grp_5 24.84
19 c Grp_5 101.45
20 d Grp_5 10500
# 虽然看上去还是数值,但已经不是数字类型了
# 而是不同的因子了,这样就可以对不同的因子赋予不同的颜色了
> is.numeric(data_m$value)
[1] FALSE
> is.factor(data_m$value)
[1] TRUE
break_v
#[1] 8.301 24.84 101.45 2794.35 5.718 10500
#18 Levels: 5.718 8.301 10.884 13.467 16.05 24.84 33.63 42.42 51.21 … 10500
# 产生对应数目的颜色
gradientC=c('green','yellow','red')
col <- colorRampPalette(gradientC)(length(break_v))
col
#[1] "#00FF00" "#66FF00" "#CCFF00" "#FFCB00" "#FF6500" "#FF0000"
p <- ggplot(data_m, aes(x=variable,y=ID)) + xlab("samples") + ylab(NULL) + theme_bw() + theme(panel.grid.major = element_blank()) + theme(legend.key=element_blank()) + theme(axis.text.x=element_text(angle=45,hjust=1, vjust=1)) + geom_tile(aes(fill=value))
# 与上面不同的地方,使用的是scale_fill_manual逐个赋值
p <- p + scale_fill_manual(values=col)
ggsave(p, filename="heatmap_nonlinear.pdf", width=8, height=12, units=c("cm"),colormodel="srgb")
调整行的顺序或列
如果想保持图中每一行的顺序与输入的数据框一致,需要设置因子的水平。这也是ggplot2中调整图例或横纵轴字符顺序的常用方式。
data_rowname <- rownames(data)
data_rowname <- as.vector(rownames(data))
data_rownames <- rev(data_rowname)
data_log_m$ID <- factor(data_log_m$ID, levels=data_rownames, ordered=T)
p <- ggplot(data_log_m, aes(x=variable,y=ID)) + xlab(NULL) + ylab(NULL) + theme_bw() + theme(panel.grid.major = element_blank()) + theme(legend.key=element_blank()) + theme(axis.text.x=element_text(angle=45,hjust=1, vjust=1)) + theme(legend.position="top") + geom_tile(aes(fill=value)) + scale_fill_gradient(low = "white", high = "red")
ggsave(p, filename="heatmap_log.pdf", width=8, height=12, units=c("cm"),colormodel="srgb")

 浙公网安备 33010602011771号
浙公网安备 33010602011771号