
文献阅读 | Optimized sample selection for cost-efficient long-read population sequencing
Ranallo-Benavidez T R, Lemmon Z, Soyk S, et al. Optimized sample selection for cost-efficient long-read population sequencing[J]. Genome Research, 2021, 31(5): 910-918. https://www.genome.org/cgi/doi/10.1101/gr.264879.120.
当使用低分辨率方法(如芯片、外显子捕获、短读长WGS等)对大型群体进行基因分型时,对其中少数具有代表性的材料进行重测序、尤其是长读长测序在群体遗传学研究中愈发重要。为了选取更具代表性的、将被用于长读长测序材料,该文章中提出了SVCollector以优化选择。
近年来越来越清楚的情况是,结构变异 (SV) 在所有生物的进化、疾病和生物学的许多其他方面发挥着关键作用。长读长测序(如 Pacific Bioscience (PacBio) 和 Oxford Nanopore Technologies )可提供更高的灵敏度和更低的错误率,但其较高的成本阻碍了在大型测序研究中的广泛应用。另一个与大型群体相关的问题是如何从基于短读段中有效地验证大量 SV。PCR/Sanger 测序等传统方法成本高昂且劳动强度大,需要仔细考虑变异和样本以验证进一步研究。
默认情况下,最佳排名力求在固定数量的样本中尽可能多地捕获遗传种群多样性。基于该方法所选的样本将包括最常见的变异以及尽可能多的稀有和私有变异。或者,它可以通过按等位基因频率对变异进行加权来优化选择,这进一步丰富了群体中的常见变异类型。样本选择的朴素方法包括随机选择或选择单独具有最多变体的样本。这些方法并不考虑变异可能在被选择的多个样本间共享的事实。
相反,SVCollector 使用优化的贪心算法来识别一个样本集合,该集合共同囊括尽可能多的变异。因此,SVCollector 允许以更具成本效益的方式来验证大量常见的 SV,以及改进的重测序方法来发现最初被短读测序遗漏的 SV。在分析中,SVCollector 报告每个所选个体存在的不同变体的累积数量。通过外推到更大的基因组集合,SVCollector 估计需要测序的个体数量,以获得特定种群的总多样性的给定部分。
SVCollector 有两种主要的排序模式:topN 和贪心(图 1),以及一个简单的随机选择模型。对于 topN 模式,它按照样本包含的变异数量的顺序选择样本,而不管这些变异是否与其他样本共享。在默认的贪婪模式下,SVCollector 会找到一个最佳样本子集,这些样本共同包含最大数量的不同变异。
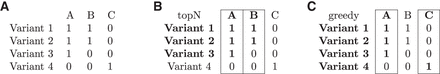
作者根据模拟数据和两个大型短读长测序项目评估了 SVCollector 的结果,项目分别涉及 2504 和 3024 个样本。对于每个队列,作者都选择了大小为 100 个不同样本的最佳集合。通过使用 SVCollector,识别的个体跨越所有亚群,而朴素的 topN 方法将选择集中在几个亚群中。对于所有队列,运行时间和内存要求都是最低的。例如,对于超过 66,555 个不同 SV 的 2504 个样本的 1000 Genomes Project VCF 文件,SVCollector 使用 1.7 MB 内存在 67 秒内计算了前 100 个样本。每种模式都有相似的运行时间和 RAM 要求。
整数线性规划 (ILP) 可以作为贪心启发式方法准确性的benchmark使用。在人类群体上,作者发现贪心算法明显优于TopN算法,并且仅比 ILP 解决方案略差 74 个 SV,同时贪心方法需要仅运行几秒钟。作者还发现,平衡随机选择在这些数据上的表现比统一随机选择差。
默认情况下,SVCollector 会最大化不同变异的计数,而不考虑变异的等位基因频率。这通常会导致识别结果中稀有或私有变异的富集,并导致常见变异捕获增加。但是,SVCollector 也可以在考虑等位基因频率的模式下运行。在这种模式下,SVCollector 也使用贪心方法,但通过根据观察到的等位基因频率对变异进行加权来优化群体中的常见变异。但测试表明,即使在等位基因频率模式下,SVCollector 也会在所有亚群中选择具有代表性的样本选择。
为了研究 SNV 和 SV 之间的关系,特别是衡量 SNV 调用是否可以用作 SV 多样性的近似值,作者使用了 1000 Genomes Project 数据并比较了三种不同的方法来挑选 100 个个体的样本,以优化涵盖的 SV 总数。总的来说,作者发现 SVCollector 在优化样本选择以最大化不同 SV 的数量方面是有效的,即使在没有 SV 调用的情况下也是如此。
此外,SVCollector 创建了一个种群多样性诊断图,其中y轴是所选样本的变异累积计数,x轴是样本数。这些 SVCollector 曲线(当使用贪婪模式生成时)使我们能够可视化随着个体添加而增加的累积变体数量的速率。作者发现这些曲线可以通过幂律分布很好地建模(对于SNV和SV均如此)。将数学模型拟合到这些曲线的主要优势在于,该模型可以外推到更多样本,以估计存在的总体特定群体多样性。然后可以使用这些外推来确定泛基因组的开放程度,并确定需要对多少个个体进行测序才能获得至少两个个体共享的给定比例的变异、完全捕获单个个体独有的私有变异需要对每个个体进行测序。
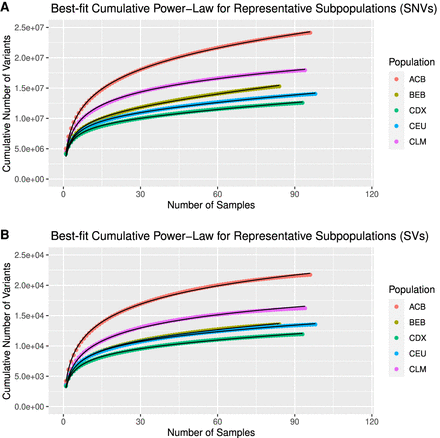
为了用不同数量的样本测试这些外推的稳健性,作者利用 1000 Genomes Project 数据进行了研究,并发现增加样本量可以提高外推法的准确性。最终通过最适的幂律曲线的外推,作者发现需要对更多的个体进行测序(尤其是非洲后裔)以充分捕捉人类变异的多样性。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号