文献阅读 | A haplotype-led approach to increase the precision of wheat breeding
Brinton, J., Ramirez-Gonzalez, R.H., Simmonds, J. et al. A haplotype-led approach to increase the precision of wheat breeding. Commun Biol 3, 712 (2020). https://doi.org/10.1038/s42003-020-01413-2
本文是10+基因组项目的一个子项目,依托于10+基因组项目(doi: 10.1038/s41586-020-2961-x)获得的基因组组装,作者寻找了小麦的单倍型模式,并构建了网站(http://www.crop-haplotypes.com/)以供查看。
单倍型的获取是通过序列间的两两比较得到的。作者采用99.99%作为两段序列属于同一单倍型的阈值,0.01%的容错率是为了兼容assembly上N和测序错误的影响,由于基于NUCmer的方法不允许在五个scaffold级装配之间进行直接比较,因此作者使用了基于滑动窗口方法的互补方法。
作者还提到,几乎相同的序列(<99.99%序列同一性)的存在与过去10,000年中小麦单倍型的预期序列差异一致(99.968%),而具有<99.5%序列同一性的物理间隔为从更遥远的野生近缘属小麦基因渗入一致的。
用于获取单倍型的小麦
10+项目的9个基因组级别的组装(代表9个品系):ArinaLrFor, Jagger, Julius, Lancer, Landmark, Mace, Norin61, Stanley, SY-Mattis
中国春Chinese Spring RefSev1.0 assembly
10+项目的5个scaffold-level assemblies(代表cultivars):Cadenza, Claire, Paragon, Robigus and Weebill
所有15个品种中,13个认为是cultivars,中国春是1900s初取自中国的地方种,ArinaLrFor来自栽培种Arina
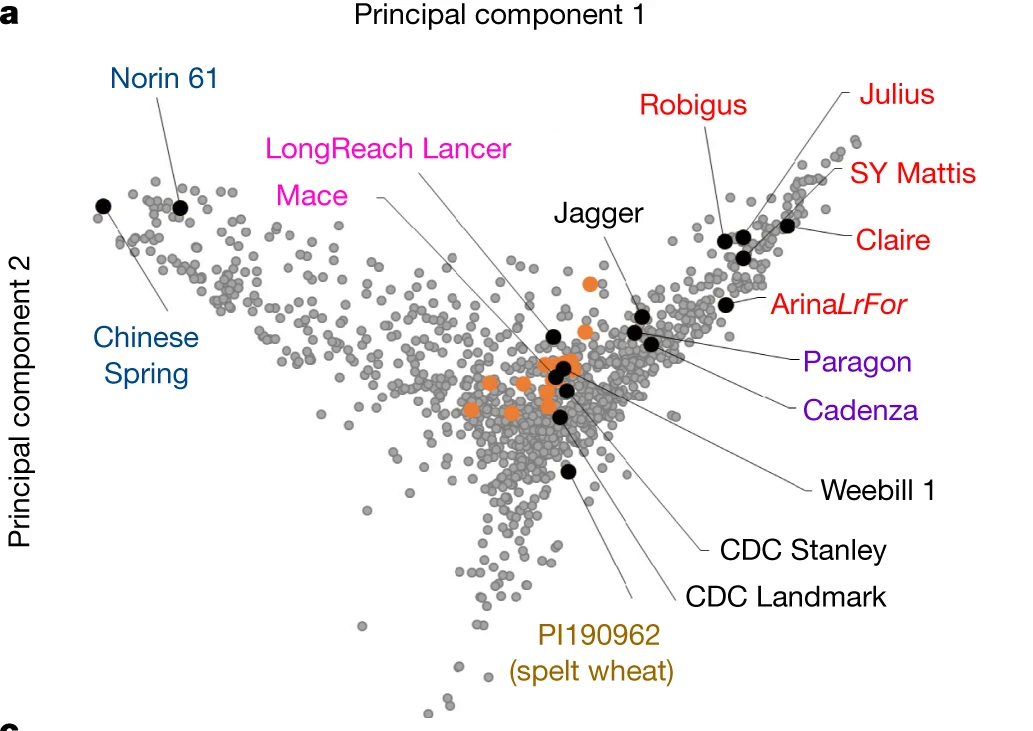
(上图自10+项目,展示了10+中组装的几个基因组所属品种在多样性空间中的位置)
单倍型获取
作者使用了NUCmer和BLASTn(基于基因的pairwise alignments)进行染色体级别的比对。
Nucmer alignments:作者使用NUCmer生成了全部15个品种间全染色体的两两比对,且不包括scaffold-level assemblies间的比较。对于染色体级别的组装,作者使用samtools-1.9 faidx提取出了单个染色体,并使用the NUCmer program from MUMmer-3.23进行比对,并采用了–mum选项来启用“anchor matches”(在reference and query中都是唯一的,以减少重复引起的匹配数)。接着,作者对生成的原始delta文件进行了过滤(options -l 20,000 (minimum length of alignment 20,000 bp), -r and -q (as recommended for one-to-one mapping of reference to query—this leaves only alignments that form the longest consistent set for the reference and query))。通过-l参数,作者去掉了小麦中非中性逆转座子之间的较短比对(shorter alignments between non-syntenic retrotransposons)(其中位数大小为9584bp)。对于scaffold-level assemblies,作者仅将其与chromosome-level进行了比对。
为了得到haplotype blocks,作者首先计算每个比对的同一性百分比,然后采用了按染色体上的位置,以5Mb大小分bin。手工结果表明,alignment sets间的gap主要由序列中的N造成,而非对小于20000bp的比对结果进行过滤造成的。在染色体级装配体的5-Mbp bin中比对的序列中位数为3,848,154 bp(即bin宽度的76.9%)。序列同一性≥99.99%(即差异小于万分之一,不采用100%是为了兼容一些技术错误如序列中的N和测序错误)(中位值)的“条带”被认为是同一状态的,即同为一个单倍型。接着,具有相同单倍型的bin会被缝合到一起。
作者还采用了2.5Mb和1Mb大小计算了bin。
BLASTn是用于对Nucmer比对进行补充。在这一环节进行了所有assemblies之间的成对比较。这一环节是对GFF annotation RefSeqv1.1中得到的基因± 2000 bp区间进行两两比较。对于染色体级装配,我们仅保留与预期染色体一致的基因投影。对于支架(Cadenza,Claire,Paragon,Robigus和Weebill)中的装配,我们根据预计基因的起源将每个支架分配给染色体。然后,我们保持基因与预期染色体一致。最后,我们滤除了预期染色体中具有多个投影的基因。
Nucmer与BLASTn的合并:为了生成最终的全基因组单倍型模块,我们合并了从NUCmer和BLAST方法调用的模块,去除了已经存在于NUCmer结果,且bin大小小于NUCmer的所有BLAST块。作者总共使用5 Mbp的bin识别了4485个成对的单倍体基因组块,使用2.5 Mbp的bin识别了7578个,使用1 Mbp识别了17693个
可视化服务器
http://www.crop-haplotypes.com/
作者使用Ruby进行后台开发,MySQL进行数据存储,D3进行绘图。
图中采用了RefSeqv1.1 genes作为所有assemblies的共同因素进行了坐标转换。
重要农艺性状基因与高度保守的单倍型分布
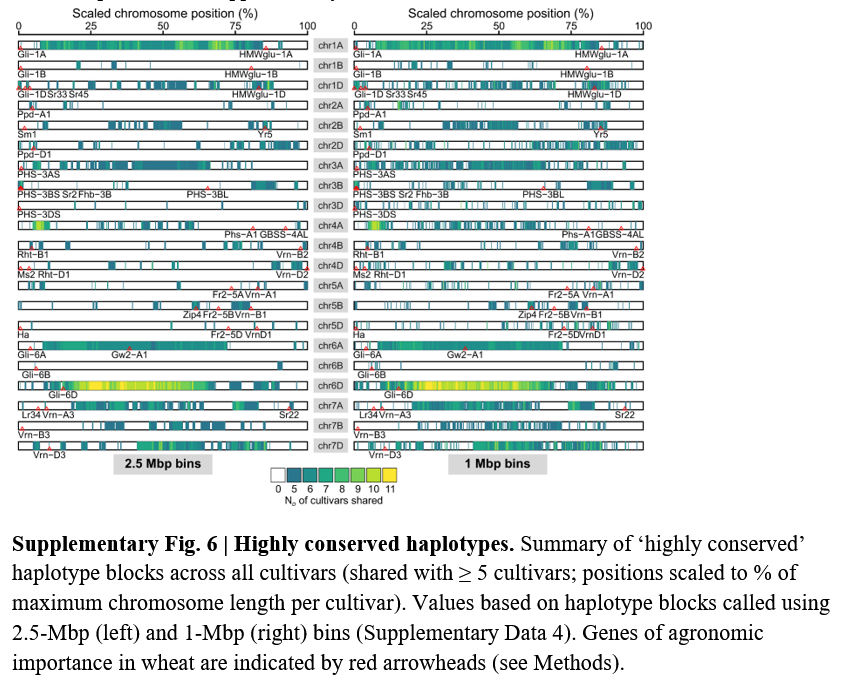
6A重组体和现场试验
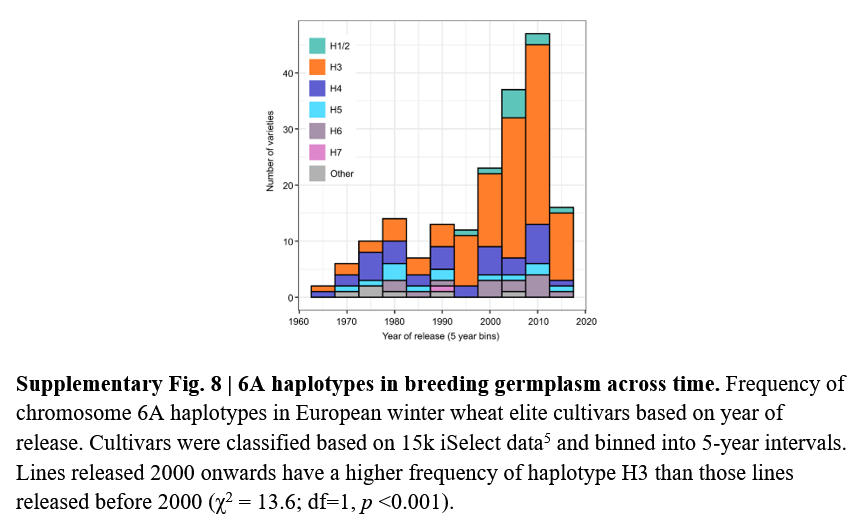
作者开发了一套初始的6A重组自交系(RIL),最终结果支持了这些单体型可以重组的想法。
几个指标
https://blog.csdn.net/index20001/article/details/77651028


 浙公网安备 33010602011771号
浙公网安备 33010602011771号