文献阅读 | Genetic Diversity, Pedigree Relationships, and A Haplotype-Based DNA Fingerprinting System of Red Bayberry Cultivars
Wu B, Zhong Y, Wu Q, et al. Genetic Diversity, Pedigree Relationships, and A Haplotype-Based DNA Fingerprinting System of Red Bayberry Cultivars[J]. Frontiers in Plant Science, 2020, 11: 1394.
本研究针对杨梅展开。杨梅是一种二倍体植物,雌雄异体。其基因组很小,约323 Mbp,且最近有三个draft genomes发表。为了开展对杨梅的遗传关系分析、筛选核心基因型、开发杨梅DNA指纹图谱,作者开展了本研究。
在单倍型重建环节,作者采用了DnaSP v6.12.03中实现的贝叶斯推断(PHASE算法)进行,并在每个片段上都运行了10000次马尔可夫链蒙特卡罗(MCMC)迭代。
杨梅群体的变异检测采用了NGS。
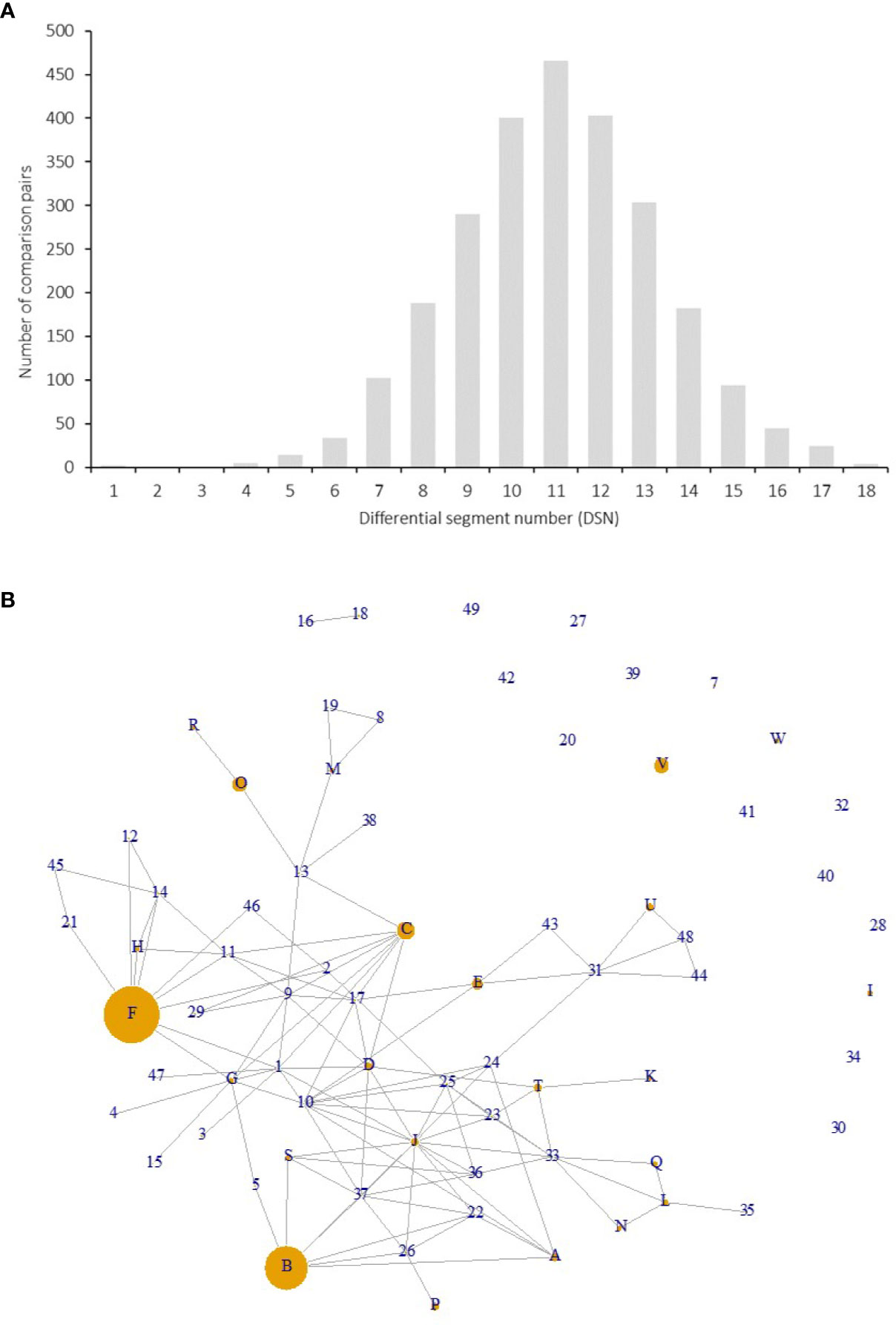
下图为基因型1-20在染色体不同segment上不同单倍型的指纹图

通过SEG1-19上72个基因型的等位基因共享分析推定了亲子关系。
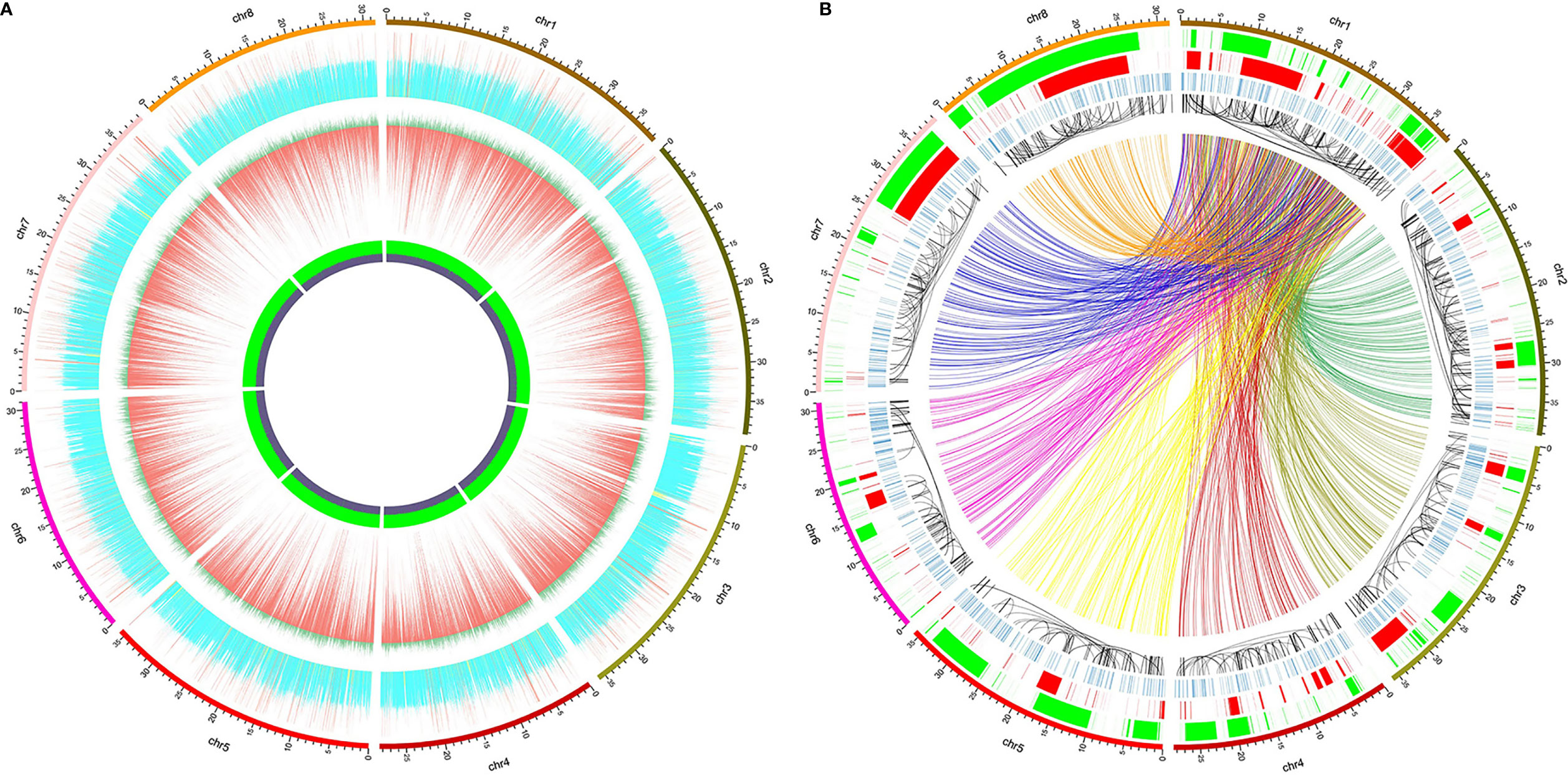
对“临海早大梅”的从头组装
在作者研究之初,尚无杨梅基因组发表。为了获得全基因组变异发现中需要的基因组草图,作者采用了SOAPdenovo2对“临海早大梅”进行了从头组装。构建contigs and scaffolds过程中构建了500bp和10kbp两种文库,并设置参数kmer为55-85、保留其他参数为原始参数。使用QUAST 4.6.0对不同kmer下组装的结果进行质量检测,并发现kmer=79有最大的N50.这一组装被用于后续使用。
全基因组变异检测
使用BWA v0.7.17将红色杨梅DNA混合物和“临海早大梅”(500 bp文库)的测序读图映射到雌性红色杨梅参考基因组(GCA_003952965.1) (Jia et al., 2019)。使用SAMtools v1.9将获取的SAM文件转换为排序的BAM格式。Bcftools v1.9使用多等位基因调用模型调用了小变异体(包括SNP和<50 bp indels)。Bedtools v2.28.0可输出整个参考基因组中连续10 kbp窗口中的测序深度和GC含量。
使用Manta v1.6.0检测到全基因组SV 。调用了五种不同类型的SV,包括BND(易位转折点),INV(反转),INS(相对于参考序列插入≥50 bp的新序列),DUP(重复)和DEL(缺失≥50 bp)。
使用Circos v0.69-8绘制了显示小变异体,SV和其他信息在参考基因组中分布的圆形图。
基因分型、单倍型构建
作者在“临海早大梅”assembly的SEG1-19中选择了15个 (not including the four duplicated segments SEG7, 11, 14, and 15) 使用Primer3设计引物。在相同条件下,使用引物对SEG1-19(补充表5)对141个部分进行PCR扩增。使用R包SangerseqR v1.22.0对从Sanger测序获得的色谱文件进行基因分型。杂合基因座以fasta格式输出为IPUAC歧义碱基,并使用MUSCLE v3.8.1551(Edgar,2004)进行多重比对。然后使用 SNP-sites v2.5.1从多个序列比对中输出所有变体和基因型。
下图展示了SEG1-16在参考基因组的pseudo-chromosomes上的分布

使用DnaSP v6.12.03中实现的贝叶斯推断(PHASE算法)在SEG1-19上重建了单倍型。使用基于单倍型的基因型,通过PowerMarker v3.25获得了单体型多样性,杂合性和PIC。通过成对比较基因型(每个重复的基因型仅保留一个基因型)来计算DSN(Pair-wise differential segment numbers)。
基因型聚类和核心集选择
使用简单匹配方法以1,000个引导程序计算72个基因型之间的遗传距离,然后通过DARwin v6.0.021(http://darwin.cirad.fr/)进行分层聚类(UPGMA)和NJ聚类(1,000个引导程序)。使用SNP上基因型的串联序列,在MEGA X中进行了1,000个UPGMA聚类的引导(Kumar等,2018)。主成分分析(PCoA)由DARwin v6.0.021在遗传距离上进行。
对于核心集基因型选择,Core Hunter v3.2.1(R包版本)(Beukelaer等人,2018)选择了2至34种不同的基因型,并将等位基因覆盖率作为评估指标。该方法要求选择的基因型覆盖所有片段上可能的最大数目的等位基因。基因型数目的上限设置为34,因为等位基因覆盖率已经达到100%。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号