文献阅读 | HaploGrouper: A generalized approach to haplogroup classification
Jagadeesan A, Ebenesersdóttir S S, Guðmundsdóttir V B, et al. HaploGrouper: A generalized approach to haplogroup classification[J]. Bioinformatics, 2020.
作者开发了一种基于已知系统树将单倍型分为单倍群(haplogroups)的通用软件。该软件的典型使用案例是将单倍群编号分配给人线粒体DNA(mtDNA)或Y染色体上的单倍型。现有的最先进的单倍群分型软件通常是“硬接线的”(hard-wired),只能处理人类的mtDNA或Y染色体单倍型。
系统发育学是进化生物学和系统学中不可缺少的工具。系统发育树一旦构建,就具有广泛的应用,其中分类是一个重要的应用。在人类中,所有活着的人类男性的Y染色体可以追溯到一个共同的祖先。同样,所有人类的mtDNA都可以追溯到一个共同的母系祖先。在这些和其他实例中,连接现存单倍型与共同祖先的分支模式反映了变异的时间顺序累积。最广泛使用的代表人类mtDNA和Y染色体多样性的系统发生树是Phylotree和International Society of Genetic Genealogy (ISOGG)’s Y tree。在这样的系统进化树中,每个分支都由至少一个将节点(单倍型)与其父节点区分开来的变异来定义(即此处的系统发育树是一个由变异信息描述的决策树,决策结果是单倍群标签)。为了便于说明这些树中特定单倍型或分支的位置,许多物种的单倍体系统(haploid systems)都采用了分支系统命名法,即用一套命名系统给各分支命名。这些具有不同名称的分支通常被称为单倍型群(haplogroups)。
当使用了能够充分刻画物种中单倍型变异特征的系统发育树时,该物种采样的大多数单倍型都应能定位到树中的已有分支。定位工作可以通过比较树上层次有序的变异和观察到的单倍型的等位基因状态来实现。为在人类mtDNA中对单倍型组进行分类、标记,已有的方法有motif matching、phylogenetic weighted ranking systems等。同时,树遍历算法(Tree traversal algorithms)被用于为Y染色体单倍型分配单倍型标签。
作者发布的HaploGrouper软件,实现了一个简单的单倍组分类的通用算法。它适用于任何具有已知的、由变异(variants)划分的树来描述单倍型(群)的单倍型系统,其中树的各个节点具有单倍群标签(编号)。该算法通过对系统进化树中每个节点与单倍型的等位基因状态的兼容性(compatibility)进行打分,来为观察到的单倍型分配单倍群标签。该软件从VCF文件中读取基因型数据,并将树和定义其分支的变异读取为两个简单的文本文件。
在描述系统发育树的两个文件中:
- 第一个文件描述了系统发育关系,其中列出了每个节点(每行一个)以及父节点。
- 第二个文件列出了树中的变异。这个文件中的每一行都包含一个变异名(大概是SNP名/编号之类的)、一个与之相关的节点(代表突变所在的分支)、物理位置,以及祖先和派生的等位基因。
运算过程:
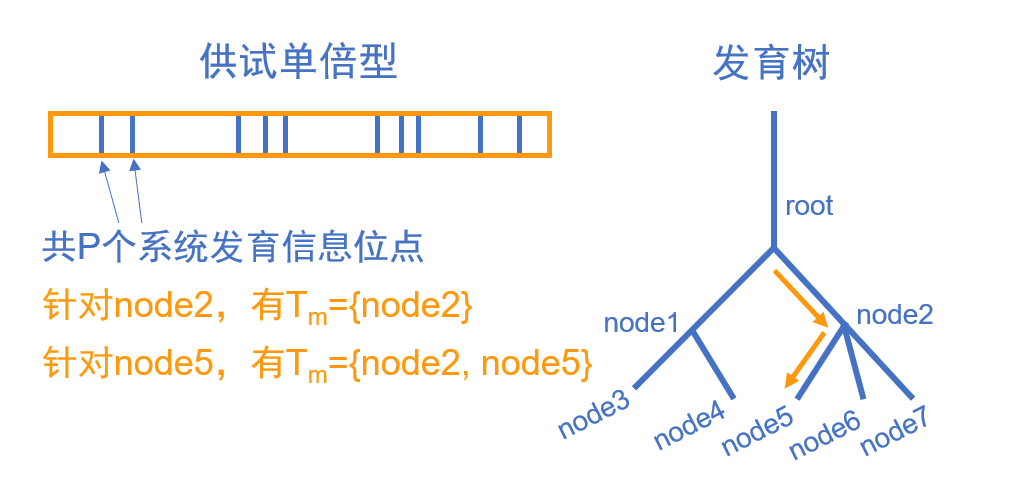
- 已知tree上记录了\(P\)个位点上的\(N\)个变异,且树上共有\(M\)个节点。
- 遍历供试单倍型上的所有系统发育信息位点(共\(P\)个),对树上的每个节点\(m\)进行打分。分数包括\(F_m\)和\(A_m\)。通常,软件会使每个系统发育信息位点对分数权重相同。
- \(F_m\)指系统发育信息位点上与当前节点一致的allele数量。考虑了位置变异率信息作为权重的\(F_m\)定义为\(F_m=\sum_{i=1}^{P}\sum_{j=1}^{k_i}d_{ij}w_i\)
- 其中\(k_i\)是第\(i\)个位点上的不同变异数量;\(w_i\)是第\(i\)个位点上的权重;\(d_{ij}\)是一个0或1的数,当测试单倍型的等位基因状态与树的第\(i\)个位置的第\(j\)个多态性的衍生等位基因匹配时,取值为1;当它们不匹配时,取值为0。
- 此处估计认为每个节点或许对应多个系统发育信息位点,每个位点对应多种allele
- 其中\(k_i\)是第\(i\)个位点上的不同变异数量;\(w_i\)是第\(i\)个位点上的权重;\(d_{ij}\)是一个0或1的数,当测试单倍型的等位基因状态与树的第\(i\)个位置的第\(j\)个多态性的衍生等位基因匹配时,取值为1;当它们不匹配时,取值为0。
- \(A_m\)指供试单倍型与当前节点不一致的allele数量(即与祖先一致的),定义为\(A_m=\sum_{i=1}^{P}\sum_{j=1}^{k_i}a_{ij}w_i\)
- 其中\(a_{ij}\)是一个0或1的数,当测试单倍型的等位基因状态与树的第\(i\)个位置的第\(j\)个多态性的祖先等位基因匹配时,取值为1;当它们不匹配时,取值为0。
- \(F_m\)指系统发育信息位点上与当前节点一致的allele数量。考虑了位置变异率信息作为权重的\(F_m\)定义为\(F_m=\sum_{i=1}^{P}\sum_{j=1}^{k_i}d_{ij}w_i\)
- 树上的每个节点\(m\)都处在一个通向祖先节点的路径\(T_m\)上,并直至树根(\(m=0\)),即\(T_m=\{0, ..., m\}\)。对每个节点\(m\)的总分\(FT_m\)和\(AT_m\)可分别通过对其祖先节点的\(F_m\)和\(A_m\)加和得到。
- 即,\(FT_m=\sum_{i \in T_m}^{}F_i\)
- \(AT_m=\sum_{i \in T_m}^{}A_i\)
- 当树上所有节点的总分都计算完毕后,下一步是计算每个节点的网络分数\(DT_m=FT_m-AT_m\)
- 最匹配供试单倍型的标签节点满足\(F_m>0\)且\(DT_m=DT_{max}\)。如果对一个单倍型分配了1个以上标签,则选择\(AT_m\)最小的节点。如果没有满足的节点,则选择竞争节点最近的共同祖先节点(作者将这种处理认为是一种保守的处理,认为这个单倍型不能被树充分描述)。
(即该方法通过打分,在系统发育决策树上寻找与供试单倍型最接近的决策)
作者将本软件与针对人类mtDNA单倍型的HaploGrep和针对人类Y染色体单倍型的yHaplo进行了对比。
为了HaploGrep和HaploGrouper的相对准确性,作者比较了271个个体与PhyloTree中给出的个体的单倍群分配。 HaploGrouper的结果与PhyloTree一致,共266人(98%),而HaploGrep 263的结果(97%)相一致(请参阅补充文本)。 分支差异的量度用于对通过两种方法获得的单倍群标签的不一致性进行打分(请参阅补充文本)。 当将两者均与Phylotree进行比较时,Haplogrouper的平均分支差异低于Haplogrep。
与yHaplo相比,HaploGrouper通过考虑缺失的衍生等位基因产生更为保守的结果。两种方法之间的其余四个差异是由于默认参数设置导致了yHaplo的树遍历算法过早终止。因此,当yHaplo在特定节点上遇到三个或更多个祖先等位基因时,它将停止探索进一步的向下路径

 浙公网安备 33010602011771号
浙公网安备 33010602011771号