文献阅读 | Systematic evaluation of error rates and causes in short samples in next-generation sequencing
Pfeiffer, F., Gröber, C., Blank, M. et al. Systematic evaluation of error rates and causes in short samples in next-generation sequencing. Sci Rep 8, 10950 (2018). https://doi.org/10.1038/s41598-018-29325-6
作者对Illumina测序仪得到的读段进行了分析,分析了错误产生的原因,总结为样本准备阶段的索引PCR对于观察到的错误率没有影响(尽管传统上将PCR视为是错误率增大的主要因素之一)。此外,尽管有软件的修正,作者仍观察到了持续的pre-phasing作用,且通过移除短序列(shortened sequences)可以消除这一效应。作者还得出了平均错误率为0.24±0.06%每碱基,以及变异序列的百分比为6.4±1.24%。5'端和3'端的恒定区(如体外选择程序中用到的引物结合位点)似乎对变异频率没有影响。考虑到phasing effects和其他测序问题在不同设备和个体的选择间存在差异,作者建议所有的NGS用户进行错误率和错误类型的分析,以改善数据质量。
NGS技术本身的错误分析
NGS技术中,最常用的是合成测序法(sequencing-by-synthesis)。据报道,该法的平均错误率在每核苷酸0.1%,其中大多数是单核苷酸替换。此外,该技术自身会产生一些错误:
- 颜色或激光串扰
- 相邻的簇之间的串扰
- phasing同步错误
- 荧光亮度弱
彩色串扰是由用于读出掺入碱基的不同荧光团之间的激发光谱和发射光谱的叠加而成的。尽管可以进行矫正,出于相同的原因,相邻簇之间的串扰依然是一个问题。
Phasing描述了两个现象,二者会导致单个序列与簇的其他部分不同步:
- 如果在一个循环中由于flow-cell未充分冲洗、甚至在终止剂被除去后由于未经结合的核苷酸仍被保留下来,而掺入了两个或多个核苷酸,就会导致pre-phasing的发生。
- Post-phasing是由于终止剂的不完全去除,而导致序列落后于簇的其余部分
下图展示了phasing错误的产生。

完全不可移动的终止剂以及对DNA链的激光损伤,会导致在一个簇中被测序的序列数量减少,进而使其荧光读数变暗。
base calling软件如Bustard包含了phasing事件的修正功能(通过假定phasing速率恒定?[1])。还有一些其他修正方法,如将附近的核苷酸纳入考虑,和在逐个运行(run-by-run)的基础上调整算法(例如,在串扰中加入循环变化[2])等。然而,作者在通过发表的软件进行phasing错误的修正中,发现尽管优于于Bustard,但完美读段的百分比仍不到77%,远低于作者通过删除缩短的序列而获得的平均94%的比例。作者认为,如果读段长度已知,则可以尝试此方法。
除上述类型之外,还有会有样本准备和测序中,PCR错误导致的变异。通过对双端测序等方法的重叠区间的研究,可以通过拒绝双链上不互补的碱基的方式降低错误率。
测序中产生的、或上述其他原因导致的的变异,可以通过index或barcode进行密切追踪。此外,单个序列的质量评估已经变得非常关键,以至于可以使用算法来确定感兴趣数据集的Phred分数的合理cut-off值[^3]。
作者的研究表明,phasing错误是初始误差率的主要因素。通过忽略变短的序列,可以排除掉被错误定相的序列,并得出每碱基0.25%的真实错误率。
与已发布的Illumina测序仪错误率相比,作者观察到的错误率处于较低范围。这可能是由于排除了缩短的序列而排除了因phasing而产生的序列。即使在省略缩短序列之前,平均错误率(无C12_EdU)也为1.56±0.81%,尽管已处于较高值,但仍符合发布的值。
样本制备过程
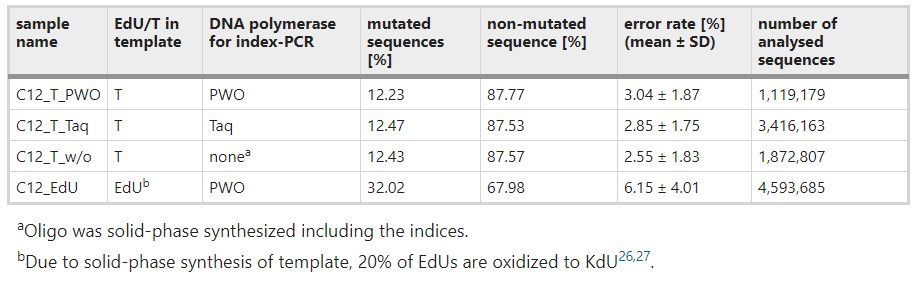
表格展示了不同样本制备方法的影响
下图a-c展示了所有三个样本每个位置的突变频率。四个原始核苷酸(original nucleotides)的平均突变频率如d图所示。e图显示了将原始核苷酸转化为真实核苷酸(the original nucleotide was converted into the denoted one)的平均突变频率。
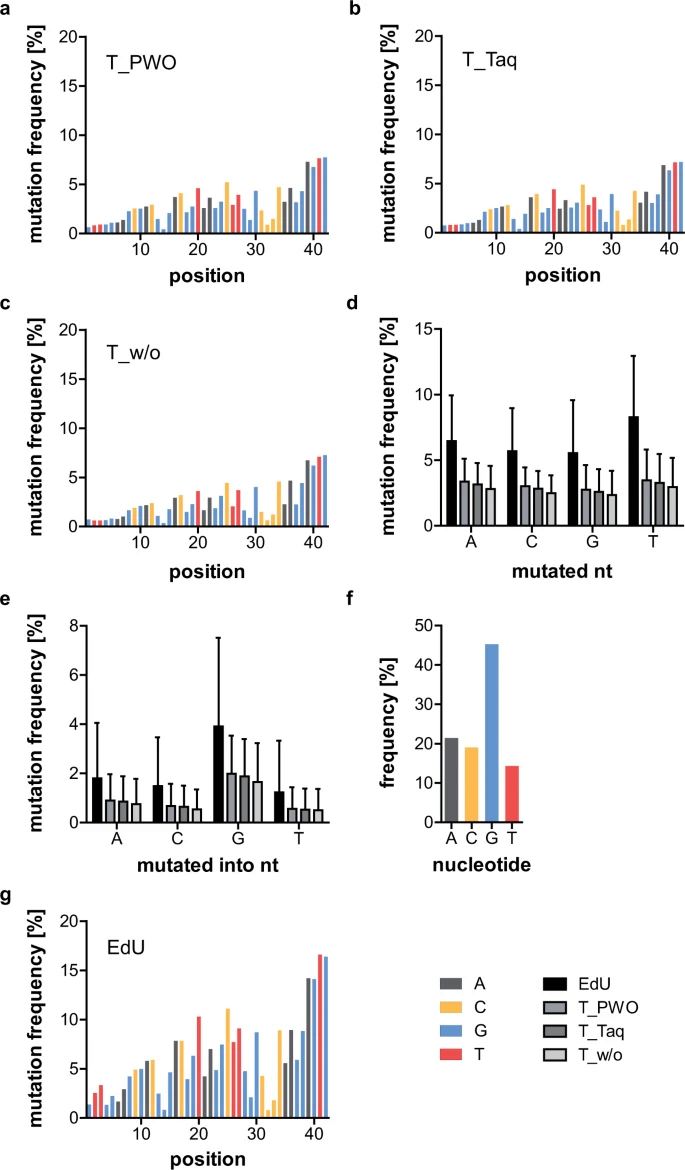
上图e中展示的平均突变频率被发现能够反映原始序列分布(f图)。为了研究这种相关性,作者设计了有重复随机区间的序列。实验表明,在所有测试样本中,后续核苷酸的突变频率随着可识别的连续核苷酸而稳定增加(from about 65 to 85%),且与核苷酸顺序无关。
测序数据的可重复性以及引物结合位点对突变率的影响
作者发现,引物结合位点似乎对错误率没有明显的影响。此外,的实验表明,来自SELEX-like libraries和序列的NGS数据看起来具有很好的可重复性。
其他参考文献
[^1] Kao, W. C., Stevens, K. & Song, Y. S. BayesCall: A model-based base-calling algorithm for high-throughput short-read sequencing. Genome Res. 19, 1884–1895, https://doi.org/10.1101/gr.095299.109 (2009).
[^2] Massingham, T. & Goldman, N. All Your Base: a fast and accurate probabilistic approach to base calling. Genome Biol. 13, R13, https://doi.org/10.1186/gb-2012-13-2-r13 (2012).
[^3] Liao, P., Satten, G. A. & Hu, Y. J. PhredEM: a phred-score-informed genotype-calling approach for next-generation sequencing studies. Genet. Epidemiol. 41, 375–387, https://doi.org/10.1002/gepi.22048 (2017).

 浙公网安备 33010602011771号
浙公网安备 33010602011771号