wig bigwig bedgraph 文件格式
测序数据分析中涉及的常用格式:
测序得到的是带有质量值的碱基序列(fastq格式),参考基因组是(fasta格式),用比对工具把fastq格式的序列比对到对应的fasta格式的参考基因序列,就可以产生sam格式的比对文件。把sam格式的文本文件压缩成二进制bam文件可以节省空间,如果对参考基因组上面的各个区段标记它们的性质,比如哪些区域是外显子、内含子、UTR等等,这就是gtf/gff格式。如果只是为了单纯描述某个基因组区域,就是bed格式文件,记录染色体号以及起始终止坐标,正负链即可。如果是记录某些位点或者区域碱基的变异,就是vcf文件格式。
fasta 基因组序列数据
fastq (测序数据) → SAM/BAM (比对) → gff/gtf (描述基因组上的结构:坐标&类型)→ Bigwig/Wiggle (信号强度,测序深度,覆盖度) → bed (描述坐标)→ vcf (突变信息)
链接:https://blog.csdn.net/qq_22253901/article/details/120543713
UCSC规定的这几个文件格式(Wiggle、bigWig和bedgraph),是为了追踪参考基因组的各个区域的覆盖度、测序深度。这些定义好的文件,可以无缝连接到UCSC的Genome Browser工具里面进行可视化!

Wiggle:简写为wig,表示基因组上一个区域的信号,可以上传至UCSC上进行可视化。Wig是一种比较老的格式,展示连续值的数据,比如GC百分比,转录组数据等。Wig数据的元素大小必须是一样的。如果数据大小不一样,应该使用bedGraph格式,如果数据过大,就转换为bigWig。
BigWig:简写为bw,是wig格式文件的二进制压缩版本,可在基因组浏览器中进行可视化,是UCSC推荐的一种格式。BigWig文件是由原始的Wig格式通过wigToBigWig工具转换过来的。
链接:https://blog.csdn.net/qq_22253901/article/details/120543713
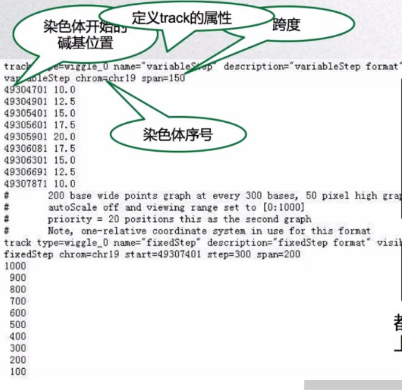
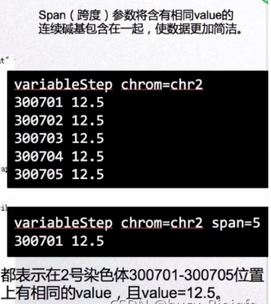
Wig文件主要由两部分格式组成:variableStep format和fixedStep format。variableStep format以一个声明开始,明确了染色体的序号,跨度(span)。后面跟两列数据,染色体开始的碱基位置,数据的值value(可以理解为覆盖度)。span参数可以将含有相同value的连续碱基包含在一起,使数据更加简洁。如图,variableStep format span=150,包含的第一行数据49304701 10.0表示49304701-49304850有相同的value,为10.0。
链接:https://blog.csdn.net/qq_22253901/article/details/120543713
http://genome.ucsc.edu/goldenPath/help/examples/wiggleExample.txt

BedGraph格式
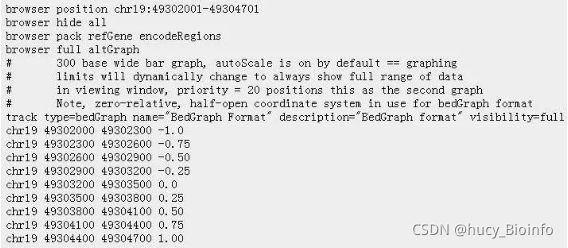
BedGraph格式文件,它是BED文件的扩展,是4列的BED格式,但是需要添加UCSC的Genome Browser工具里面显示的属性,一般就定义有限的几个属性即可。
BedGraph,它的trace type和Wig文件很像,不过后面的数据和bed文件很类似,后面的四列分别表示染色体序号,起始位置,结束位置和value值。
链接:https://blog.csdn.net/qq_22253901/article/details/120543713
REF
https://blog.csdn.net/qq_22253901/article/details/120543713
https://www.freesion.com/article/44761417825/
https://www.jianshu.com/p/abb0add3d459/
https://genome.ucsc.edu/goldenpath/help/wiggle.html
WIG格式(.wig)
Wig的数据包括track line和data line。
- track line 定义了track的属性,比如track type=wiggle_0,指定track为Wig track。
- data line 主要由两部分格式组成,variableStep format和fixedStep format。variableStep format以一个声明开始,明确了染色体的序号,跨度(span)。后面跟两列数据,染色体开始的碱基位置,数据的值value(可以理解为覆盖度)。span参数可以将含有相同value的连续碱基包含在一起,使数据更加简洁。第二部分为fixedStep format, 由声明和单列数据组成。声明部分和variableStep format中各变量的意义一样。wig中的value值可以是整数,实数,正数或者负数。只有指定的位置有value值,没有制定的位置则没有value,且不会在UCSU Genome Browser中作出图。
BIGWIG格式(.bw,.bigwig)
wig或bedGraph的索引二进制文件,也就是可以由这两种文件转换得到
在处理大型数据集时,bigWig文件的显示性能比常规的wig文件快得多
链接:https://www.jianshu.com/p/e2af596c7c35
wig示例文件:
https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSM1027287&format=file&file=GSM1027287%5FUW%2ECD19%5FPrimary%5FCells%2EH3K27ac%2ERO%5F01701%2EHistone%2EDS21712%2Ewig%2Egz
1 head GSM1027287_UW.CD19_Primary_Cells.H3K27ac.RO_01701.Histone.DS21712.wig 2 track type=wiggle_0 name="UW.CD19_Primary_Cells.H3K27ac.RO_01701.Histone.DS21712" description="UW.CD19_Primary_Cells.H3K27ac.RO_01701.Histone.DS21712" visibility=full color=20,150,20 3 fixedStep chrom=chr1 start=9001 step=20 span=20 4 0 5 0 6 0 7 0 8 0 9 0 10 0 11 0
1 fixedStep chrom=chr1 start=783001 step=20 span=20 2 0 3 0 4 0 5 0 6 0 7 0 8 0 9 0 10 0 11 0 12 0 13 0 14 1 15 1 16 1 17 1 18 1 19 1 20 1 21 1 22 1 23 1 24 1 25 0 26 0 27 0 28 0 29 0 30 0 31 0 32 0 33 0 34 0 35 0 36 0 37 1 38 1 39 1 40 2 41 2 42 2 43 2 44 2 45 2 46 2 47 2 48 2 49 2 50 2 51 1
Wiggle Track ASCII Text Format (.wig)
Wiggle
v.(使)扭动,摆动
n.扭动,摆动;波浪形线
Wiggle files and its bedgraph variant allow you to plot quantitative data as either shades of color (dense mode) or bars of varying height (full and pack mode) on the genome. Both are text files that are easy to create, but need to be converted for actual use by the genome browser.
The bedGraph format is a very similar format for sparse data or data that contains elements of varying size. bedGraph can also be converted to compressed/indexed binary bigWig files. If you have other data to show in addition to the quantitative data, e.g. data you want to show on mouseover or when the user clicks the feature (like GWAS data), you should have a look at bigBed files with the "lollipop" type (contact us for more info). For a list of all possible formats for graphing, see the following wiki page.
Text files in wiggle format can be uploaded as custom tracks as-is to UCSC where they are compressed and stored for some time. But we recommand that you convert them on your own computer to the binary bigWig storage format. You then copy bigWig files onto your own webserver and they are referenced in custom tracks or track hubs via their URL.
Unlike bigWig binar files, wiggle ASCII text files can be uploaded as custom tracks onto our server. After the upload, wiggle data is compressed and stored internally in 128 unique bins. This compression means that there is a minor loss of precision when data is exported from a wiggle track (i.e., with output format "data points" or "bed format" within the Table Browser). For custom tracks, use the bedGraph format if it is important to retain exact data when exporting. However, the size of all custom tracks is limited. For these reasons, we recommend always converting wiggle files to the bigWig storage format and reference these from your custom tracks or track hubs via their URL.
General structure
Wiggle format is line-oriented. For wiggle custom tracks, the first line must be a track definition line (i.e., track type=wiggle_0), which designates the track as a wiggle track and adds a number of options for controlling the default display. For conversion to bigWig, the most common use case, this line must not be present.
Wiggle format is composed of declaration lines and data lines, and require a separate wiggle track definition line. There are two options for formatting wiggle data: variableStep and fixedStep. These formats were developed to allow the file to be written as compactly as possible.
variableStep format
This format is used for data with irregular intervals between new data points, and is the more commonly used wiggle format. After the wiggle track definition line, variableStep begins with a declaration line and is followed by two columns containing chromosome positions and data values:
variableStep chrom=chrN
[span=windowSize]
chromStartA dataValueA
chromStartB dataValueB
... etc ... ... etc ...The declaration line starts with the word variableStep and is followed by a specification for a chromosome. The optional span parameter (default: span=1) allows data composed of contiguous runs of bases with the same data value to be specified more succinctly. The span begins at each chromosome position specified and indicates the number of bases that data value should cover. For example, this variableStep specification:
variableStep chrom=chr2
300701 12.5
300702 12.5
300703 12.5
300704 12.5
300705 12.5 is equivalent to:
variableStep chrom=chr2 span=5
300701 12.5
Both versions display a value of 12.5 at position 300701-300705 on chromosome 2.
Caution about sparse variableStep data: The wiggle format was designed for quickly displaying data that is quite dense. The variableStep format, in particular, becomes very inefficient when there are only a few data points per 1,024 bases. If variableStep data points (i.e., chromStarts) are greater than about 100 bases apart, it is advisable to use BedGraph format.
fixedStep format
This format is used for data with regular intervals between new data values and is the more compact wiggle format. After the wiggle track definition line, fixedStep begins with a declaration line and is followed by a single column of data values:
fixedStep chrom=chrN
start=position step=stepInterval
[span=windowSize]
dataValue1
dataValue2
... etc ...The declaration line starts with the word fixedStep and includes specifications for chromosome, start coordinate, and step size. The span specification has the same meaning as in variableStep format. For example, this fixedStep specification:
fixedStep chrom=chr3 start=400601 step=100
11
22
33
displays the values 11, 22, and 33 as single-base regions on chromosome 3 at positions 400601, 400701, and 400801, respectively. Adding span=5 to the declaration line:
fixedStep chrom=chr3 start=400601 step=100 span=5
11
22
33 causes the values 11, 22, and 33 to be displayed as 5-base regions on chromosome 3 at positions 400601-400605, 400701-400705, and 400801-400805, respectively.
Note that for both variableStep and fixedStep formats, the same span must be used throughout the dataset. If no span is specified, the default span of 1 is used. As the name suggests, fixedStep wiggles require the same size step throughout the dataset. If not specified, a step size of 1 is used.
Data values
Wiggle track data values can be integer or real, positive or negative values. Only positions specified have data. Positions not specified do not have data and will not be graphed. All positions specified in the input data must be in numerical order. NaN values are not supported by the browser and, if included, may have unforeseen effects.
1-start coordinate system in use for variableStep and fixedStep
The bedGraph format, like all BED-based formats and most file formats used by UCSC, use "0-start, half-open" coordinates, but the wiggle ASCII text format for variableStep and fixedStep data uses "1-start, fully-closed" coordinates. Wiggle (variableStep and fixedStep) is the only format defined by UCSC that uses a 1-based format, for historical reasons. For example, for a chromosome of length N, the first position is 1 and the last position is N. For more information, see:
- BigWig and BigBed: enabling browsing of large distributed datasets (Bioinformatics)
- Database/browser start coordinates differ by 1 base (Genome Browser FAQ)
- The UCSC Genome Browser Coordinate Counting Systems (Genome Browser blog)
Parameters for custom wiggle track definition lines
All options are placed in a single line separated by spaces (line breaks are inserted in the following example to facilitate readability):
track type=wiggle_0 name=track_label
description=center_label
visibility=display_mode color=r,g,b
altColor=r,g,b priority=priority
autoScale=on|off alwaysZero=on|off
gridDefault=on|off
maxHeightPixels=max:default:min
graphType=bar|points
viewLimits=lower:upper
yLineMark=real-value yLineOnOff=on|off
windowingFunction=mean+whiskers|maximum|mean|minimum
smoothingWindow=off|2-16The track type with version is REQUIRED, and it currently must be wiggle_0:
type wiggle_0The remaining values are OPTIONAL:
name <trackLabel> # default is "User Track"
description <centerLabel> # default is "User Supplied Track"
visibility <full|dense|hide> # default is hide
color <RRR,GGG,BBB> # default is 255,255,255
altColor <RRR,GGG,BBB> # default is 128,128,128
priority <N> # default is 100For a list of additional options, see the bigWig format page. Note that bigWig files created from bedGraph cannot be converted to wiggle using bigWigToWig and instead will be reverted back to their original bedGraph format. Also, for tracks using altColor with the windowing function "mean+whiskers" the shading of colors will be impacted with lighter shades for values within a standard deviation around the mean, most noticeable when zoomed out and average calculations are taking place.
Examples
This example specifies 19 separate data points in two tracks (the first is name="variableStep" and the second is name="fixedStep" where priority= orders them) in the region chr19:49,304,200-49,310,700. To view this example as a custom track in the Genome Browser, copy the text and paste it into the "Add Custom Tracks" text box.
browser position chr19:49304200-49310700
browser hide all
# 150 base wide bar graph at arbitrarily spaced positions,
# threshold line drawn at y=11.76
# autoScale off viewing range set to [0:25]
# priority = 10 positions this as the first graph
# Note, one-relative coordinate system in use for this format
track type=wiggle_0 name="variableStep" description="variableStep format" visibility=full autoScale=off viewLimits=0.0:25.0 color=50,150,255 yLineMark=11.76 yLineOnOff=on priority=10
variableStep chrom=chr19 span=150
49304701 10.0
49304901 12.5
49305401 15.0
49305601 17.5
49305901 20.0
49306081 17.5
49306301 15.0
49306691 12.5
49307871 10.0
# 200 base wide points graph at every 300 bases, 50 pixel high graph
# autoScale off and viewing range set to [0:1000]
# priority = 20 positions this as the second graph
# Note, one-relative coordinate system in use for this format
track type=wiggle_0 name="fixedStep" description="fixedStep format" visibility=full autoScale=off viewLimits=0:1000 color=0,200,100 maxHeightPixels=100:50:20 graphType=points priority=20
fixedStep chrom=chr19 start=49307401 step=300 span=200
1000
900
800
700
600
500
400
300
200
100
https://genome.ucsc.edu/goldenpath/help/bigWig.html
bigWig Track Format
The bigWig format is useful for dense, continuous data that will be displayed in the Genome Browser as a graph. BigWig files are created from wiggle (wig) type files using the program wigToBigWig. Alternatively, bigWig files can be created from bedGraph files, using the program bedGraphToBigWig.
The bigWig files are in an indexed binary format. The main advantage of this format is that only those portions of the file needed to display a particular region are transferred to the Genome Browser server. Because of this, bigWig files have considerably faster display performance than regular wiggle files when working with large data sets. The bigWig file remains on your local web-accessible server (http, https or ftp), not on the UCSC server, and only the portion needed for the currently displayed chromosomal position is locally cached as a "sparse file". If you do not have access to a web-accessible server and need hosting space for your bigWig files, please see the Hosting section of the Track Hub Help documentation.
Wiggle data must be continuous and consist of equally sized elements. If your data is sparse or contains elements of varying sizes, use the bedGraph format instead of the wiggle format. If you have a very large bedGraph data set, you can convert it to the bigWig format using the bedGraphToBigWig program. For details, see Example #3 below. Refer to this wiki page for help in selecting the graphing track data format most appropriate for the type of data you have.
Note that the wigToBigWig utility uses a substantial amount of memory: approximately 50% more memory than that of the uncompressed wiggle input file. While running the wigToBigWig utility, we recommend that you monitor the system's memory usage with the top command. The bedGraphToBigWig utility uses about 25% more RAM than the uncompressed bedGraph input file.
Creating a bigWig track
To create a bigWig track from a wiggle file, follow these steps:
Step 1. Create a wig format file following the directions here. When converting a wig file to a bigWig file, you are limited to one track of data in your input file; therefore, you must create a separate wig file for each data track.
Step 2. Remove any existing "track" or "browser" lines from your wig file so that it contains only data.
Step 3. Download the wigToBigWig program from the binary utilities directory.
Step 4. Use the fetchChromSizes script from the same directory to create the chrom.sizes file for the UCSC database with which you are working (e.g., hg19). If the assembly genNom is hosted by UCSC, chrom.sizes can be a URL like: http://hgdownload.soe.ucsc.edu/goldenPath/genNom/bigZips/genNom.chrom.sizes
Step 5. Use the wigToBigWig utility to create the bigWig file from your wig file:
wigToBigWig input.wig chrom.sizes myBigWig.bwNote that the wigToBigWig program also accepts gzipped wig input files.
Step 6. Move the newly created bigWig file (myBigWig.bw) to a web-accessible http, https, or ftp location.
Step 7. If the file name ends with a .bigWig or .bw suffix, you can paste the URL directly into the custom track management page, click "submit" and view the file as a track in the Genome Browser. By default, the file name will be used to name the track. To configure the track label or other visualization options, you must create a track line, as shown in Step 8.
Step 8. Construct a custom track using a single track line. The most basic version of the track line will look something like this:
track type=bigWig name="My Big Wig" description="A Graph of Data from My Lab" bigDataUrl=http://myorg.edu/mylab/myBigWig.bwPaste the custom track line into the text box on the custom track management page.
bigWig custom track lines can have several optional parameters, including:
autoScale <on|off> # default is on alwaysZero <on|off> # default is off gridDefault <on|off> # default is off maxHeightPixels <max:default:min> # default is 128:128:11 graphType <bar|points> # default is bar viewLimits <lower:upper> # default is range found in data viewLimitsMax <lower:upper> # suggested bounds of viewLimits, but not enforced yLineMark <real-value> # default is 0.0 yLineOnOff <on|off> # default is off windowingFunction <mean+whiskers|maximum|mean|minimum> # default is maximum, mean+whiskers is recommended smoothingWindow <off|[2-16]> # default is off transformFunc <NONE|LOG> # default is NONE
For further information on custom bigWig track settings, see the Track Database Definition Document. For more information on how bigWig settings are used in native Genome Browser tracks, see the Configuring graph-based tracks page.
Examples
Example #1
In this example, you create a bigWig custom track using an existing bigWig file on the UCSC http server. The file contains data that spans chromosome 21 on the hg19 assembly.
To create a custom track using this bigWig file:
- Paste the URL
http://genome.ucsc.edu/goldenPath/help/examples/bigWigExample.bwonto the custom track management page for the human assembly hg19 (Feb. 2009). - Click the "submit" button.
- On the next page that displays, click the "chr21" link in the custom track listing to view the bigWig track at position chr21:33,031,597-33,041,570 in the Genome Browser.
Alternatively, you can customize the track display by including track and browser lines that define certain parameters:
- Construct a track line that references the bigWigExample.bw file:
track type=bigWig name="Example One" description="A bigWig file" bigDataUrl=http://genome.ucsc.edu/goldenPath/help/examples/bigWigExample.bw - Include the following browser line to ensure that the custom track opens at the correct position:
browser position chr21:33,031,597-33,041,570 - Paste the browser and track lines onto the custom track management page for the human assembly hg19 (Feb. 2009), click the "submit" button, then click the "chr21" link in the custom track listing to view the bigWig track in the Genome Browser.
Example #2
In this example, you will create your own bigWig file from an existing wiggle file.
- Save this wiggle file to your computer (Steps 1 and 2 in Creating a bigWig format track, above).
- Save the file hg19.chrom.sizes to your computer. This file contains the chrom.sizes for the human (hg19) assembly (Step 4, above).
- Download the
wigToBigWigutility (step 3, above). - Run the utility to create the bigWig output file (step 5, above):
wigToBigWig wigVarStepExample.gz hg19.chrom.sizes myBigWig.bw - Place the newly created bigWig file (myBigWig.bw) on a web-accessible server (step 6, above).
- Paste the URL of the bigWig file into the custom track entry form, or construct a track line that points to your bigWig file (step 7, above).
- Create the custom track on the human assembly hg19 (Feb. 2009), and view it in the Genome Browser (step 8, above). Note that the original wiggle file spans only chromosome 21.
Example #3
To create a bigWig track from a bedGraph file, follow these steps:
- Create a bedGraph format file following the directions here. When converting a bedGraph file to a bigWig file, you are limited to one track of data in your input file; therefore, you must create a separate bedGraph file for each data track.
- Remove any existing track or browser lines from your bedGraph file so that it contains only data.
- Download the
bedGraphToBigWigprogram from the binary utilities directory. - Use the
fetchChromSizesscript from the same directory to create the chrom.sizes file for the UCSC database with which you are working (e.g., hg19). If the assemblygenNomis hosted by UCSC, chrom.sizes can be a URL likehttp://hgdownload.soe.ucsc.edu/goldenPath/genNom/bigZips/genNom.chrom.sizes - Use the
bedGraphToBigWigutility to create a bigWig file from your bedGraph file:
(Note that the bedGraphToBigWig program DOES NOT accept gzipped bedGraph input files.)bedGraphToBigWig in.bedGraph chrom.sizes myBigWig.bw - Move the newly created bigWig file (
myBigWig.bw) to a web-accessible http, https, or ftp location. - Paste the URL into the custom track entry form or construct a custom track using a single track line.
- Paste the custom track line into the text box on the custom track management page.
Example #4
In this example, we will display a bigWig with a dynamic sequence logo (Motif Logo) using logo=on.
- Construct a track line that references the bigWigExample.bw file:
track type=bigWig logo=on name="Example dynseq" description="A bigWig file with logo=on dynseq" visibility=full autoScale=off bigDataUrl=http://genome.ucsc.edu/goldenPath/help/examples/bigWigExample.bw - Include the following browser line to ensure that the custom track opens at the correct position:
browser position chr21:33,037,000-33,037,050 - Paste the browser and track lines onto the custom track management page for the human assembly hg19 (Feb. 2009), click the "submit" button, then click the "chr21" link in the custom track listing to view the bigWig track in the Genome Browser.
You can also load the above by clicking this link.
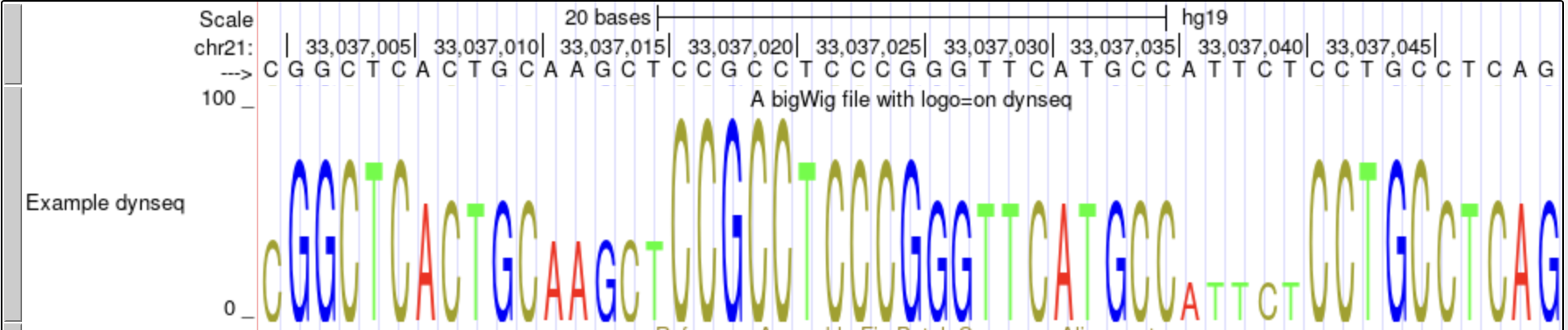
This dynseq display scales nucleotide characters by user-specified, base-resolution scores and was developed by the Kundaje Lab.
Sharing your data with others
If you would like to share your bigWig data track with a colleague, learn how to create a URL by looking at Example #6 on this page.
Extracting data from the bigWig format
Because bigWig files are indexed binary files, it can be difficult to extract data from them. UCSC has developed the following programs to assist in working with these files, available from the binary utilities directory.
bigWigToBedGraph— converts a bigWig file to ASCII bedGraph format.bigWigToWig— converts a bigWig file to wig format. Note: if a bigWig file was created from a bedGraph, bigWigToWig will revert the file back to bedGraph.bigWigSummary— extracts summary information from a bigWig file.bigWigAverageOverBed— computes the average score of a bigWig over each bed, which may have introns.bigWigInfo— prints out information about a bigWig file.
These utilities accept either file path names or URLs to files as input. As with all UCSC Genome Browser programs, simply type the program name (with no parameters) on the command line to view the usage statement.
In some cases, bigWigSummary and bigWigAverageOverBed will produce very similar results, but in other cases, the results may differ. This is due to data-handling differences between the two programs. Summary levels are used with bigWigSummary; therefore, some rounding errors and border conditions are encountered when extracting data over relatively small regions. In contrast, the bigWigAverageOverBed utility uses the actual data, which ensures the highest level of accuracy.

 浙公网安备 33010602011771号
浙公网安备 33010602011771号