顺式调控元件(Cis)在植物中的研究进展及未来的挑战
https://academic.oup.com/plcell/article/34/2/718/6433166?login=false
cis-regulatory modules (CRMs)
cis-regulatory elements (CREs)
We refer here to CREs as individual transcription factor (TF) binding sites, while CRMs are assemblies of CREs and include promoters, transcriptional enhancers, silencers, and insulator elements.
解析基因表达调控的机制,是生物学家们一直以来想要解决的基本问题。尽管在高质量序列的组装和基因组注释已经取得了重要的进展,但在全基因组范围内对影响基因表达的顺式调控元件(CRE)和顺式调控模块(CRM)的识别与表征则仍旧面临很多问题;例如找到能够用于描述不同CRE的标记物以及区分它们的活性状态等。
2021年11月22日,荷兰阿姆斯特丹大学Maike Stam团队在plant cell上发表了题为“Cis-regulatory sequences in plants: Their importance, discovery, and future challenges”的综述性论文。作者回顾了当前对CRM认识,以及描述在CRM的识别、表征和验证方面的突破性技术。与此同时比较了CRM在不同植物基因组中的分布,并讨论了包含有CRM的转座子在基因表达方面的进化作用。
CRM的重要组成部分-转录因子结合位点(TFBs)
CRMs是众多CREs的集合而与TF特异性结合的序列是真核生物基因组中表达调控的关键组成部分。在特定细胞中表达的TF并与CRM结合从而决定了CRMs的活性和最终的功能(图1)。要激活一个CRMs,首先先锋
TFs(pioneer
TFs),结合到核小体DNA上并且招募组蛋白乙酰转移酶和其他染色质重塑复合物,使得染色质的结构发生改变。随后其他的非先锋TFs和辅助因子与开放的染色质区域(ACRs)DNA结合,最终使得CRMs被完全激活。
在真核生物中,大约只有5~7%的细胞核基因编码TFs蛋白,而大多数TF-binding sites(TFBS)只有6-12 bases长度并且在基因组中广泛分布。识别有特定TF调控的序列是一个比较热门的研究领域。在体内,同一个基因家族的TFs能够识别相似的DNA序列,但是具有不同的DNA结合亲和力;因此这种基于旁系同源TF之间建立结合偏好性同样很重要。
对于大多数TFs,核小体给TF和CREs的结合提供了天然的屏障。不同于大多数的TFs,先锋TFs能够与一些inaccessible染色质上的核小体结合,使得CRMs能够招募激活或者阻遏物来激活或抑制转录。
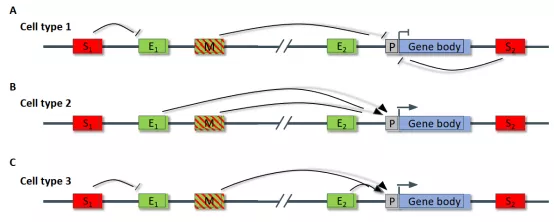
图1:CRMs在不同类型的细胞中引发不同的转录反应
S: silencer,E: enhancer, M: multifunctional sequence element, p: Promoter.
CRMs中调控元件的主要特征
CRMs中其活性状态的特点主要是由DNA和染色质特征的不同组合所描述,而启动子(promoter)和增强子(enhancer)的特征相比于沉默子(silencers)更加明确(图2)。在植物中绝大多数的CRMs出现在未甲基化区域而这些区域的甲基化往往具有组织特异性,并且在accessible 染色质区域富集。由于植物基因组中大多数区域被甲基化,包括基因区域和转录沉默的TE区域;因此植物基因组中未甲基化的区域可能包含了绝大多数的CRMs。
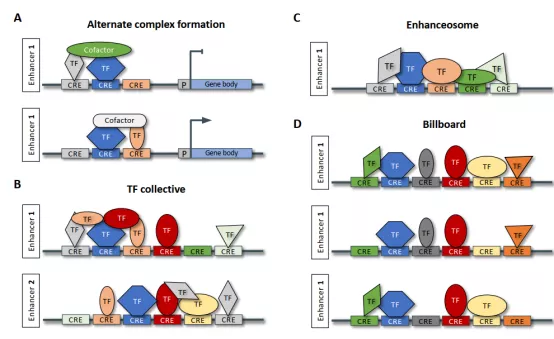
图2:CRMs模型
CRMs的几种活跃状态也已经被描述,但是并非所有的状态都被明确的定义,作者将CRMs的或与状态分为以下3种:repressed,poised 和active(图3)。通过不同的染色质修饰例如H3K27me3和HAc,使得CRMs的活性状态发生改变。例如处于poised 状态下的CRMs能够被激活或者被抑制,它们通常结合了少量的TFs,富含H3K27me3以及少量的激活性HAc组蛋白修饰。
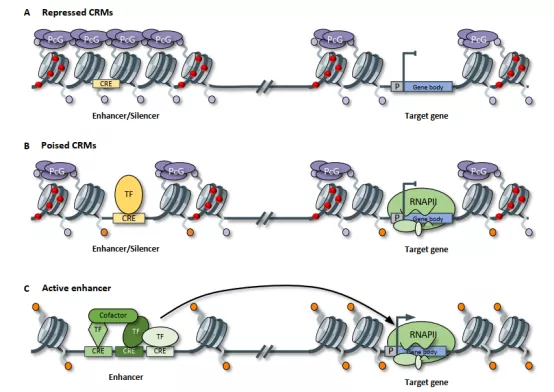
图3:不同状态下的CRMs的染色质可及性以及修饰
CRMs的识别
如何精确的定位和表针CRM以及其目标基因是一项具有挑战性的工作,作者主要讨论了两种主要途径:
1. 基于靶向方法对单个CRM进行识别与验证
2. 全基因组识别与验证
通过开发“增强子陷阱”方法用于识别和鉴定未知的CRMs。这种方法通量低、只能对单个CRMs精细鉴定且在对小基因组且易于转化的物种中非常有用,但对于大基因组的生物例如棉花、玉米等仍旧十分困难;其他的方法就是使用数量性状基因座(QTL)和精细定位方法。
高通量测序技术的出现极大的促进了在全基因组范围内识别CRMs。通过利用表观基因组的特征,进行特征组合用于识别CRMs和靶基因。DNA甲基化数据可以用于在全基因组范围内对CRMs进行预测。对于组织特异性的CRMs,可以通过DNAse-seq、ATAC-seq、MNase-seq是被特性性的ACRs区域,从而对CRMs进行表征。基因GWAS关联分析用于鉴定存在变异位点的CRMs,例如在植物中通过eQTL分析鉴定基因表达量和非编码区的关联。在玉米中,GWAS鉴定到到大于40%的表型遗传变异位于ACRs区域,这些变异很可能是由于CRMs的变异导致基因表达的改变。
大量的CRMs与它所调控的基因距离超过10Kb甚至更远,这也使得CRMs的鉴定变得更为复杂。在这方面可以利用CRMs与靶基因之间发生的物理上染色质的相互作用,这种互作通常被称为基于染色质构象捕获技术(3C)(图4)。Hi-C能够揭示全基因组范围内染色质的相互作用,尤其是对于一些基因组相对小的生物来说。而在基因组比较大的植物中,则采用了另外一种替代的方法例如ChIA-PET和Hi-ChIP分析与特定组蛋白修饰或者特定蛋白质发生相互作用的CRMs。
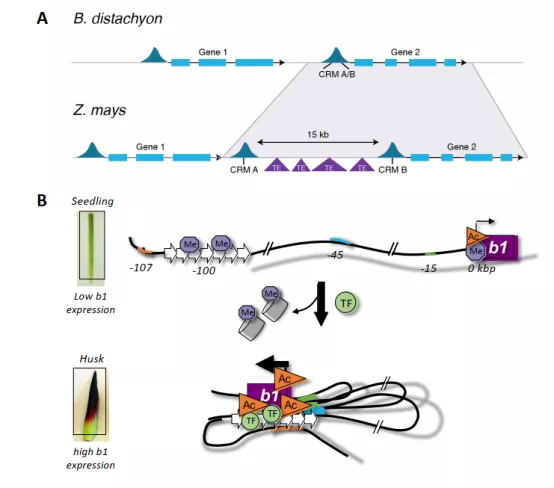
图4:远距离的CRM和染色质互作
CRMs的功能验证
对潜在的CRMs的调控活性进行验证也是一个巨大的挑战,其中的瓶颈在于转基因功能获得或者丧失检验的低通量。在这些实验中假的的CRMs通常与报告基因一起被克隆,通过报告基因的激活或者沉默用于判断CRMs的活性。另外一种方式则是通过使用RNA介导的DNA甲基化或者突变的方式使得目标CRMs失活,但这种方式需要注意的是全基因组范围内CRMs功能可能冗余,从而掩盖了突变的实际作用(图5A)。
一种高通量的验证方法例如self-transcribing active regulatory region-sequencing(STARR-seq)能够对基因组范围内的CRMs进行大规模的评估。在STARR-seq中,基因组片段被打断并添加barcode,打断后的片段被克隆到报告基因的3’UTR区域,从而创建了一个报告基因库中衍生的RNA片段表明了CRMs的活性(图5B)。
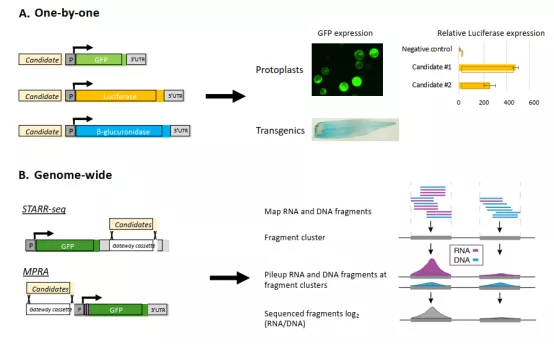
图5:CRMs的功能验证
转座子衍生的CRMs
转座子可以通过几种不同的机制影响基因的表达其中包括:直接破坏CRMs、携带沉默性的染色质标记扩散到基因的侧翼区域、为基因提供新的CRMs等。在本文中作者仅仅讨论了后者TE携带新的CRMs方式,从而调控基因的表达。在人类和玉米中大于有25%和30%的调控序列是转座子所衍生的(TE-CRMs)(图6)。由于TE-CRMs在基因组中具有移动和扩增的潜力,因此物种特异性的CRMs通常富集在TEs中。具有有利影响的TE插入可能在进化过程中被保留,导致基因调控网络的重新布线以及新的基因网络的形成,而不利影响的插入很可能被自然选择所淘汰。除此之外,与其他类型的CRMs一样,TE同样具有组织、时空特特异表达,只有在特定的组织和调节才能检测到TE-CRMs的存在。
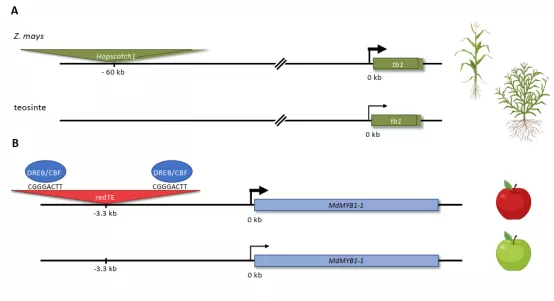
图6:TE-CRMS的实例
未来的展望与挑战
一个主要的挑战时如何鉴定植物中特定组织和条件下的CRMs,这个可能会通过新兴的单细胞测序技术来实现。虽然单细胞RNA-seq在植物中已经正在被应用,但是其他类型的单细胞技术例如ChIp-seq、Hi-C和甲基化在哺乳动物中刚刚实现,还没有完全的在植物中应用。
如何鉴定假的的增强子和沉默子以及两者相互作用共同调控基因表达;迄今为止这方面的知识仍旧是空白的。另外一个挑战则是鉴定CRMs和其互作的靶基因,尤其是大量CRMs除了调节直接靶基因外还可能调节其他基因。总的来说我们正处于发现和挖掘这些CRMs的阶段,但未来需要将更多的注意力放在如何应用这样知识,利用CRISPR诱变和自然界中已有的遗传变异推动其在作物设计育种和改良方面的应用。
REF
https://mp.weixin.qq.com/s/djwbWQifmGFam-5u6Cs_YQ

 浙公网安备 33010602011771号
浙公网安备 33010602011771号