ORR氧气电还原测试流程
 ORR测试方法、转移电子数计算、过氧化氢浓度测定
ORR测试方法、转移电子数计算、过氧化氢浓度测定
1. 测试前的准备
1.1 准备电解液
- 酸性电解液:0.5M H2SO4或0.1M HClO4
- 碱性电解液:0.1M KOH
- 中性电解液:0.1M PB(磷酸缓冲液)
注:贵金属催化剂尤其是铂基催化剂必须使用高氯酸才能获得较好的性能。
1.2 准备参比电极
- 酸性电解液:Ag/AgCl (sat. KCl) 参比电极
- 碱性电解液:Hg/HgO (1M KOH) 参比电极
- 中性电解液:Hg/Hg2Cl2 (sat. KCl) 参比电极
注:实测发现Ag/AgCl参比通常也能在0.1M KOH中长时间稳定测试,1M KOH中容易使参比的盐桥溶液变黄失效。
1.3 检查气瓶的气压水平
气压不足时及时订气。氧气的小气瓶容量8 L,压强5 MPa,以100 sccm速率使用,大约仅能用66小时。大气瓶容量40L,压强20 MPa,氧气量是小气瓶的20倍,因此如果需要进行长时间性能测试最好使用大气瓶。
准备惰性气体用于吹扫,一般氮气即可,不一定要用氩气。注意惰性气氛通气速率过慢时容易得到过高的基线。
根据亨利定律,理想气体在溶液中的溶解度与该气体在液面上方的分压成正比,因此使用空气测试时,相比使用纯氧,溶液中氧气的溶解度会降低,相应地会使反应速率降低、过电位增大。
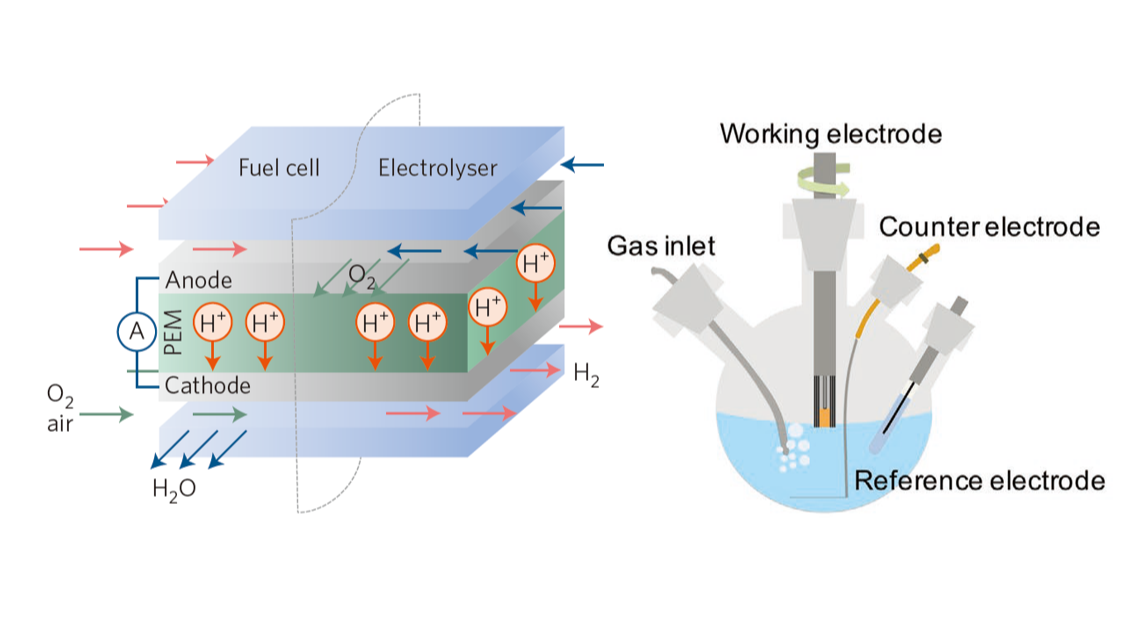
1.4 准备五口电解池
ORR测试应使用五口电解池。HER/OER常用的三口反应池开口较小,难以将曝气管插在工作电极一侧,插在对电极因扩散路径过长严重降低ORR反应极限电流。五口池中间的24磨口插入旋转圆盘电极,侧面的19磨口分别插入砂芯导气管、石墨对电极、参比电极,侧面多出一个磨口留空。
许多的测试教程中说ORR电解池应密封,上文侧面留空的磨口位应接液封,通气使用F型砂芯管,吹扫时从液面下方通气,测试时从液面上方通气。实测发现,旋转圆盘电极与中间磨口之间不知道如何密封,有资料显示似乎有密封配件存在,但是买不到。在中间磨口无法密封的情况下,侧磨口液封是没有意义的,实在介意的话直接用磨口塞堵上即可。用F型导气管从液面上方通气是一个不切实际的操作,因为反应消耗的氧气无法及时得到补充,测得的性能一定比液面下通气更差。
1.5 准备工作电极
以往可以使用普通的RDE(旋转圆盘电极),利用在不同扫速下测试的结果,用K-L法计算转移电子数。如今K-L法已被彻底淘汰,所以测试时务必使用RRDE(旋转环盘电极)。ORR电流密度较低,对测试环境的敏感度比HER更高,应该尽量在每次测试前磨好电极。如果电极没有明显的大划痕,在麂皮上用50 nm粒径的氧化铝粉抛光即可,使用前应用超纯水润湿麂皮,抛光时均匀施力将电极按在麂皮上画8字(不能画圈,否则电极会被磨歪)。磨好后先用无尘纸擦去表面大部分的氧化铝粉,在20mL样品瓶中装满乙醇,在乙醇中将电极头超声3秒左右以除去电极表面残留的抛光粉,然后用无尘纸擦干表面的乙醇,最后检查电极表面是否呈镜面反射。
倒置旋转电极装置,以200 rpm旋转时滴加浆液,然后在700rpm下旋转干燥。转速为参考值,可根据浆液实际情况相应调整。滴加量参考:纯乙醇浆液单次滴加少于5 uL,含水浆液单次少于10 uL。通常而言,即使是同样的催化剂、同一批的浆液,在电极上的负载量越大,选择性就越偏向4电子,反之负载量越小,选择性就越偏向2电子。因此,务必控制确保不同样品间的负载量相同。
工作电极表面应在电解液液面下方5~10 mm处。刚插入电极时,电极上有气泡是正常的,可以直接进行CV活化操作,通常跑8个循环后气泡就基本消失。如果仍有大气泡,应上下移动电极位置,重新浸入电解液中以尝试赶走气泡,重复跑CV直至电极没有气泡。气泡存在时极化电流远大于电极浸润在溶液中时,实验数据必须是在没有气泡的条件下测得才有可比性。
2. 测试流程
- 扫CV活化电极:参考设置:设置转速1600 rpm, 扫速0.05 V/s, 碱性条件下相对参比0.2V ~ -0.8V范围扫12圈,灵敏度10-3 A。一般来说刚放入电解液的电极会在表面糊上一个气泡或有一层气膜,需要先扫CV活化。
- RDE扫不同转速LSV计算转移电子数(已弃用的方法):参考设置:设置转速1600 rpm, 扫速0.01 V/s, 碱性条件下相对参比0.2V ~ -0.8V范围,灵敏度10-3 A。应确认电极表面无气泡、无气膜后开始测试。如有气泡,应将电极抬起到液面上面再重新浸入电解液中,如果仍存在气膜,应再扫一次CV。改变转速测LSV,用K-L曲线粗测转移电子数。转速依次取400 rpm, 625 rpm, 900 rpm, 1225 rpm, 1600 rpm, 2025 rpm, 2500 rpm。低转速注意避免气流过大导致的电流扰动;高转速应调大气流量,确保不同扫速的LSV曲线的平台间距基本相同,若各转速叠加发现高转速时间距变小则说明气流不足。
- RRDE扫LSV测转移电子数:参考设置:设置转速1600 rpm, 扫速0.01 V/s, 碱性条件下相对参比0.2V ~ -0.8V范围,灵敏度10-3 A,打开第二电极,固定电位1.2 V vs RHE,灵敏度10-4 A。一般可以用铂环电极测,碱性条件下测产过氧化氢的催化剂应使用金环电极。
- 测试EIS电化学阻抗谱(可选):参考设置:CHI工作站测试频率范围100000Hz~1Hz,其它设置保持默认,如果只需要得到未补偿电阻,测试电位可以只测开路电位,如果要对比不同材料的EIS谱,应补充在有反应发生的电位下的测试(不然理论上开路电位下EIS不应当测出半圆而只能测得未补偿电阻),建议分别测一个到达极限电流的电位和一个未到达极限电流的电位备用。
3. 转移电子数的计算方法
3.1 Koutecky-Levich法(已弃用的方法)

方程中:
j 代表实际测得的电流密度,
jK 代表动力学电流密度,
n 代表转移电子数,
F 代表法拉第常数 (F = 96485 C mol-1),
D 代表氧气在电解液中的扩散常数 (0.1M KOH中,D = 1.9×10-5 cm2 s-1),
υ 代表溶液粘度 ( υ = 0.01 cm2 s-1),
C 代表氧气的饱和浓度 (C = 1.2×10-3 mol L-1),
ω 代表旋转圆盘的转速 (单位: rpm)。
0.1M KOH中,公式常数项 0.620FD2/3υ-1/6C = 0.11,此时公式可简化为 1/j = 1/jk + (1/0.11n) * ω-1/2。取某一电位下不同转速ω对应的电流密度j,以ω-1/2为x,以1/j为y,进行线性拟合,由得到的斜率k求得转移电子数n = 1/0.11k。在每一个不同电位下都会算得一个对应的n。
目前发表论文已经基本不使用K-L法,ORR转移电子数测试以RRDE为准。
3.2 RRDE法

式中,n代表转移电子数,Idisk代表测量的盘电流,Iring代表环电流,N代表电极的电子收集率。
特别注意:此处应使用电流值,而非电流密度。使用RRDE法测试,在不同电位下,都有一个对应的转移电子数,这是正常的,并且也应当在论文中报告不同电位下的转移电子数。
4. 过氧化氢产量测试(2电子ORR催化剂适用)
4.1 H-cell测试
物品清单(供参考)
- 电解池:30 mL 高仕睿联H型电解池带循环水夹层
- 隔膜:Nafion 212 阳离子交换膜(推荐,酸碱通用)或 PK-130阴离子交换膜
- 阴极电解液:30 mL 1M KOH + 10 mM Na2EDTA(简写为EDTA,抑制H2O2在碱性条件下被金属离子催化的歧化反应)
- 阳极电解液:30 mL 2M KOH(实测1M KOH无法完成反应,泡沫镍表面糊上一层绿色氧化物层)
- 参比电极:Hg/HgO (氯化银电极无法耐受1M KOH长时间反应)
- 对电极:3 cm * 3 cm 泡沫镍(带1 cm * 1.5 cm把手,供电极夹夹持)
- 工作电极:YLS-30T 气体扩散层 1 cm * 2 cm(浆液只涂在末端的1 cm * 1 cm区域)
- 浆液:4 mg/mL * 25 uL,负载量 0.1 mg/cm2
- Pt电极夹 3只,红色鳄鱼电极夹 1只,橡皮筋
- 屏蔽箱(遮光抑制H2O2的分解)
测试方法(供参考)
- 恒电位模式: 0.4VRHE电位下电解12小时
- 恒电流模式: 100 mA恒电流电解
如果采用恒电流电解,不同催化剂之间实际上是在比较该电流密度下的法拉第效率/电子转移数的差别,因此更推荐恒电位电解,可以对比不同材料在相同电位下催化反应的速率差异。
4.2 比色法测产物浓度
- 如果材料的催化效率很高,电解液中的过氧化氢含量较高,可以使用高锰酸钾滴定法(非常准确哦),不过前期标定溶液浓度的准备比较麻烦,本科读材料专业的同学通常没有学过分析化学实验,一般也不愿意操作,所以还是用分光法吧。
- 有些材料的性能很差或者电解选用的电流密度很低,导致电解产生的过氧化氢含量极低,此时高锰酸钾滴定法通常是不现实的,因为在如此低的浓度下高锰酸钾的颜色极浅,加之高锰酸钾滴定起始时反应速率偏低,极难判定反应终点,此时也必须采用比色法。
- 比色法需要的过氧化氢浓度极低,通常为2 mg/L左右,普通的过氧化氢浓度为30%左右,需要稀释15万倍,用容量瓶需要定容2~3次,非常麻烦而且最终浓度不确切。由于过氧化氢的浓度直接决定了标准曲线的斜率,并且高浓度的过氧化氢通常含有抑制剂,因此务必购买过氧化氢标准溶液,常见浓度为1000 μg/ml。
- 工作曲线上的点通过向相同量的显色溶液中加入不同浓度梯度的过氧化氢溶液获得。切换不同样品时对比色皿的清洗可能会导致背底吸收变化,因此应当注意扣背底。
TiOSO4比色法
- 反应方程式 TiOSO4 + H2O2 + H2SO4 → H2[Ti(O2)(SO4)2] + H2O (络合反应)
- 特征吸收峰 H2[Ti(O2)(SO4)2] 405 nm(测试范围600 nm ~ 350 nm,每个样品重复三次)
- 配置钛标准液 购买的硫酸氧钛溶液是15 wt%溶液,稀释2000倍(取250 uL定容到500 mL)定容时直接加3M H2SO4预酸化,大约需要80 mL浓硫酸. 测试时每管加2 mL. 注:加入钛溶液的量固定时,其实不需要知道其确切浓度,确保足够与加入的过氧化氢反应即可.
- 配置过氧化氢 购买的标液浓度为1000 μg/ml,稀释100倍(取5 mL定容到500 mL)到10 μg/ml. 定标准曲线时每管加0~3 mL,再用去离子水加到5 mL.
- 注意事项 这一反应必须在酸性条件下进行,本方案在钛标液中预先加酸,可直接加入电解液;由于仪器限制,务必每次测试时都重定标准曲线,并尽快完成全部测试;如果不加过氧化氢的样本吸收率偏移线性区较多,被测液浓度较低时可以先加1 mL过氧化氢标准液垫高吸收度;本法中已经形成的黄色产物H2[Ti(O2)(SO4)2]吸光度是不会随时间推移减小的(保质期>2个月);可以用600 nm~590 nm作为背底数据,扣背底后极大提升线性度和重复性.
Ce4+比色法
- 反应方程式 Ce4+ + ½ H2O2 → Ce3+ + ¼ O2 + H+
- 特征吸收峰 Ce4+ 320 nm(测试范围500 nm ~ 200 nm,每个样品重复三次)
- 注意事项 Ce4+极易水解,碱性电解液务必先酸化后使用!
- 不推荐本法 此法在2e ORR文献中最常见,但是存放在10 mL塑料离心管的Ce4+吸光度随时间推移极速降低,甚至相隔10分钟就有明显区别,完全无法正常、可靠地完成测试;前期测试发现电解后得到的电解液的背底吸收很高,远高于低浓度Ce4+产生的吸收强度,无法正常完成比色法;文献显示过氧化氢本身在350 nm以下(紫外区)开始有强吸收。
Cu(phen)2+比色法
- 反应方程式 Cu(phen)2+ + ½ H2O2 → Cu(phen)+ + ¼ O2 + H+
- 特征吸收峰 Cu(phen)+ 453 nm
- 注意事项 本方法未实际测试过;文献报道此反应需要一定的反应时间才能达到最大吸光度,最好预留半小时时间。
5. 拓展阅读
- Angew论文:Fe-O4位点高效电合成过氧化氢 https://onlinelibrary.wiley.com/doi/10.1002/anie.202410123
- 摸着羊的笔记本:极限扩散电流到底是不是材料的本征性能?https://www.mozheyang.top/2019/03/31/LSV-KLeq/
- 邵阳、徐梽川AM:实验操作综述 http://doi.org/10.1002/adma.201806296
- 乔世璋ACS Catalysis:使用RRDE和金环电极评估转移电子数 http://dx.doi.org/10.1021/acscatal.6b01581
- 测试狗:旋转电极的基本原理及应用 https://www.ceshigo.com/article/10248.html

 浙公网安备 33010602011771号
浙公网安备 33010602011771号