
1、导读
分子进化的重要原理如基因进化的重复进化(gene duplication)、分子钟等(moleculer clock)都是通过研究氨基酸序列发现的。
这里之所以先介绍氨基酸进化的原因:
1)比DNA序列更加保守
2)蛋白质编码基因的DNA序列的对位排列分析上,需要氨基酸序列矫正
3)氨基酸的进化演变模型比DNA序列简单的多
本章的重要目的:介绍两个氨基酸序列间进化距离(evolutionary distance)的统计方法。
研究进化距离的意义:
1)对研究蛋白质进化非常重要
2)可用来构建系统树并估计分歧时间
在氨基酸序列方面,通常以氨基酸替代数来测定距离,但不同假设下存在不同的测度。
2、氨基酸的差异和不同氨基酸的比例
2.1)最简单的测度
两个序列间氨基酸个数差异(nd)。如果所有的氨基酸的个数相同(n),上述差异数就可以用来比较不同序列间的分歧程度。当多重序列比对时候,经常会含有插入缺失,这种情况下计算nd需要去掉插入缺失(间隔,indels),否则不同序列对间相比较时候计算出来的nd没有意义。nd:number of amino acid differences
2.2)p距离
两个序列间差异氨基酸所占的比例.即使n随着不同序列而变化,但比例值(p)依旧可以用于比较分歧程度,该距离为p距离(p为proportion的缩写)。公式为:
![]()
假设所有的氨基酸位点都是以相同的概率替代(即每个位点差异的概率都是p,相当于伯努利实验),则nd需遵循二项式分布,,因此其方差为:
![]()
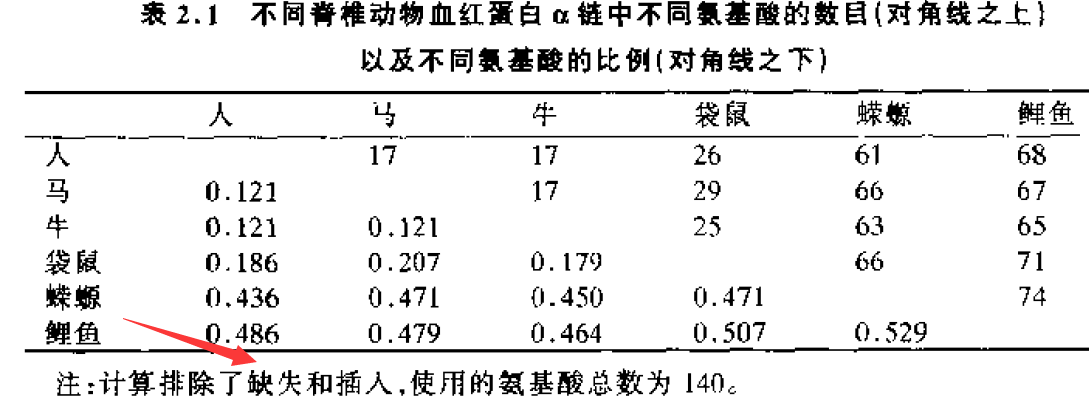
n=140,nd位于对角线上,p值位于对角线下。可以看出,物种关系远时,p值大;物种关系近时,p值小。说明:随着物种分歧时间增大,氨基酸替代数目也增加。但是p并不严格与分歧时间(t)成比列。 下面将给出解释。
3、 泊松校正和τ距离
p与t成非线性关系的原因:同一个位点氨基酸的多重替代(multiple amino acid substitution), 这导致nd偏离实际氨基酸替代数逐渐增加。更精确的估计替代数方法是是用泊松分布。
3.1、假设氨基酸替代率在每一个位点都相同
r:特定位点的氨基酸替代率,这里假设每一个位点替代率相同(不是真实情况,但次假设误差很小,除非p非常大)。t年之后,每个位点氨基酸替代数为rt.在一个给定位点氨基酸替代数k(k=0,1,2,3,4,5,6) 发生的频率遵循泊松分布。即:
![]()
因此某一位点氨基酸不变的概率为:
![]()
如果多肽链长度为n,不变氨基酸的期望为ne-rt
但在实际应用中,并不知道祖先物种的氨基酸序列,这样上述公式就不能用。因此只能对已有t年分化的两个同源序列进行比较来估计氨基酸的替代数。
由于一个氨基酸位点无替代的概率是e-rt,因而两个序列同源位点均无替代的概率是:q=(e-rt)2=e-2rt. 而q=1-p.
两个序列间每个位点氨基酸替代总数为d(d=2rt)为:d=-ln(1-p)
以![]() 代替p,可以获得d的估计值
代替p,可以获得d的估计值![]() ,即泊松校正距离,同时
,即泊松校正距离,同时![]() 的方差为:
的方差为:
![]()
分子进化研究中通常需要知道氨基酸的替代率(r),如果从其他生信手段已经知道两个序列的分化时间t, 此速率的估计值为:
![]()
注意:是2t,而不是t.因为该速率是一个进化系速率。
![]() 的方差为
的方差为![]()
另一方面如果我们得知速率r,但不清楚进化时间t,可以用下式表示:
![]()
其 方差为:
![]()
3.2 实际情况中每一个氨基酸位点的替代率并不都相同
以上所有的公式都假设的是氨基酸每个位点的替代率是相同的。然而事实并非如此,因为功能上次要的位点比功能上重要的位点常常含有更高的替代率。每个位点氨基酸替代率(k)分布的方差大于泊松方差,且次此方差近视值遵循负二项分布。若已知每个位点的氨基酸替代率(r)按照τ分布的话,每个位点氨基酸替代的观察值将按照负二项分布。因此建议不同位点的替代率都按照τ分布估计。即:
![]()
3.3 突变率和替代率
只有当突变扩展到群体中,才能使得突变参与到基因组中。这一事件称为突变再群体中的固定。一旦突变固定,群体中每一个个体都携带同样突变。再比较不同物种的氨基酸序列时候,我们主要研究已经固定到这些物种的基因组中的突变的氨基酸的演变。

即,每个基因座的基因替代率等于突变率。
在氨基酸序列数据方面,通常考虑每个氨基酸位点的替代率。如果以每年每个氨基酸位点突变率(u)来定义突变率,则每年每个位点的氨基酸替代率等于突变率。

在某些情况下:


 浙公网安备 33010602011771号
浙公网安备 33010602011771号