
1)背景
处理基因组数据中,比较基因组不同区域,例如寻找overlap等,是一种基本的且常见的问题。虽然UCSC 中‘Table Browser’或者Galaxy可以用来处理,但是当这些工具面对大的数据的时候就会显得力不从心。因此,需要一款快速、灵活的软件来批量处理数据集。
bedtools是一款用C++编写的小巧且灵活的软件来处理这些复杂的问题,可以用来比较、操作、注释bed和gff文件中的genomic features。它设计主要是在linux环境下,可以和awk、grep、sort 等实现无缝对接,且支持bam格式文件,可以将bam文件转换为bed文件,因此对于大数据处理来说非常方便。
由于在版本在不断的更新,我们将依据最初版本的参考文献给出每个示例图,让大家知道理解每个命令的含义,知道如何去学习一个软件。明白了原理,具体要到的时候,依据不同版本的帮助文档就很容易上手。
2)功能
下图是bedtool的可用操作(注意版本不同,命令可能会变,但大致功能不会改变,这里主要是展示其原始文献):虽然有些命令已经该换名字,例如fastaFromBed在后续的版本中的别名是getfasta,但是这些命令已经可以被执行,尽管后续版本不断有新的参数被加入。(在一再次注意,这里主要通过图解来说每个命令到底在做什么。不讲实际应用)

2.1)intersectBed
‘intersecting’ or ‘overlapping’指feature之间至少有1bp 的重叠:
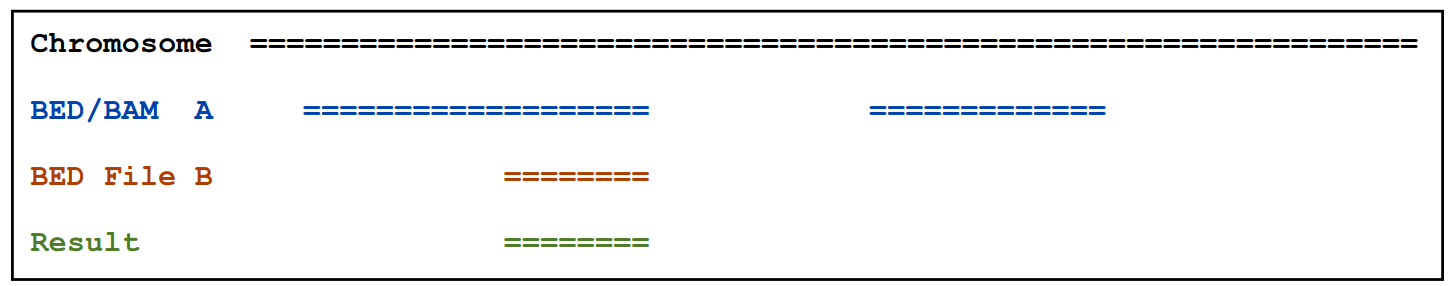
基本用法:intersectBed [OPTIONS] [-a <BED> || -abam <BAM>] -b <BED>

2.1.1)找出overlap并输出
intersectBed –a A.bed –b B.bed

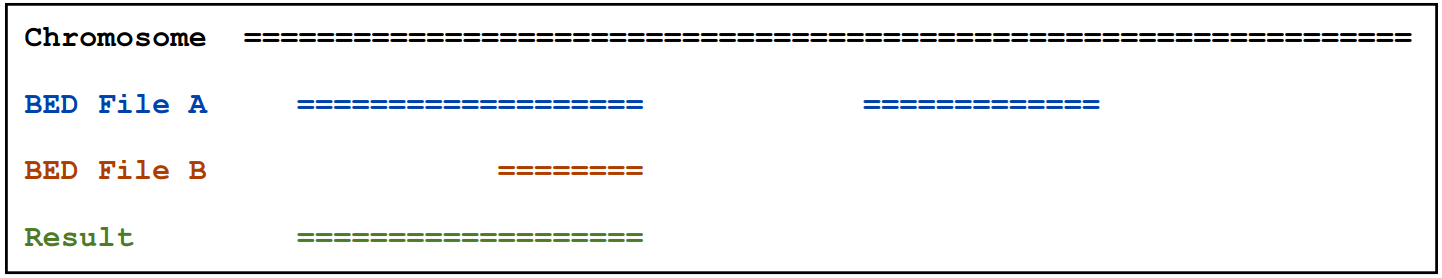
2.1.2)当有overlap输出原始的 “A” feature
intersectBed –a A.bed –b B.bed -wa


2.2)pairToBed
主要用于将BedPE feature或者paired-end BAM alignment 与另一个bed文件比较寻找overlap
基本用法:pairToBed [OPTIONS] [-a <BEDPE> || -abam <BAM>] -b <BED>
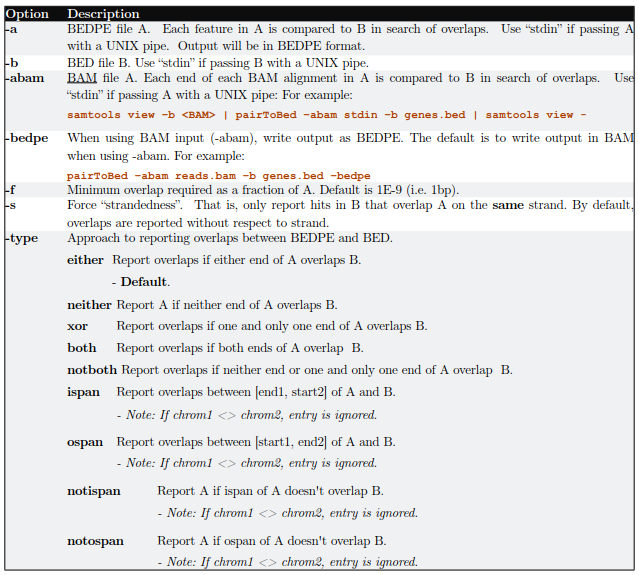
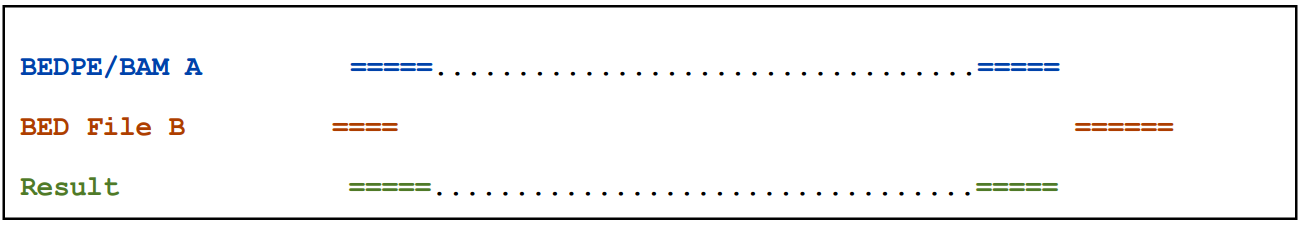
2.2.1)默认情况下,如果任何一端与B有交集则输出A(Report A if either end overlaps B.)
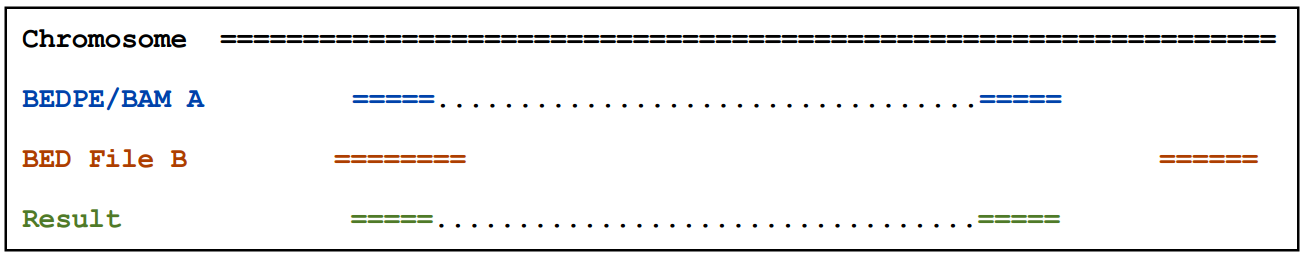
2.2.2)-type 参数可以用来控制输出
-type both: 当用该参数的时候,只有在双端都和B有交集的情况下才输出A(Report A only if both ends overlap B)

-type neither: 如果双端和B都没有交集则输出A(Report A only if neither end overlaps B.)

-type ispan: 输出A,如果A双端间区域和B有重叠(Report A if it’s “inner span” overlaps B.)
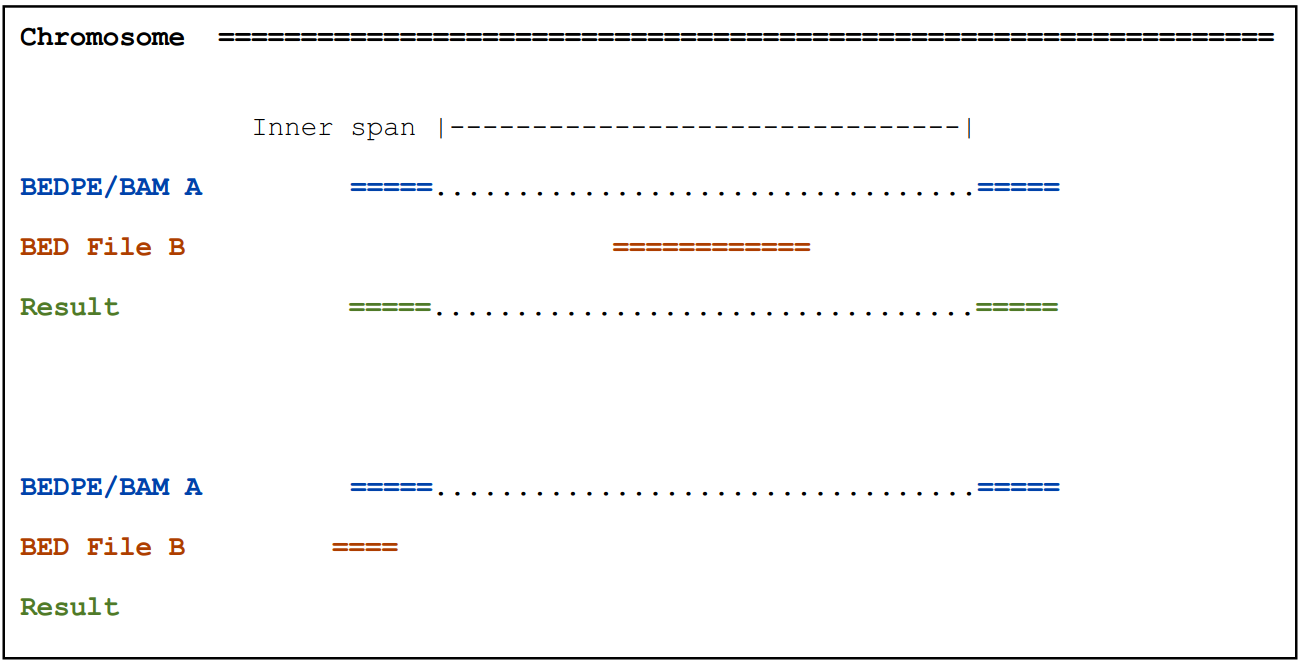
2.2.3) -f参数通过调节最少比对长度来控制输出
输出A,如果一端至少有50%区域与B重叠(eport A only at least 50% of one of the two ends is overlapped by B)
pairToBed -a A.bedpe -b B.bed -f 0.5

2.3) pairToPair
寻找两个BEDPE文件之间的重叠
基本用法:pairToPair [OPTIONS] -a <BEDPE> -b <BEDPE>

2.3.1)默认情况下输出A,如果A和B都有链信息,且A的双端和B有交集

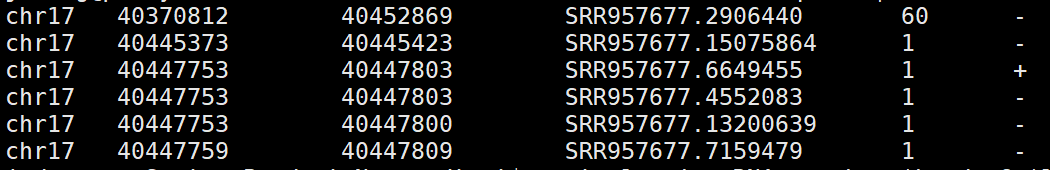
2.4)bamToBed
用来将bam格式转化为Bed格式
基本用法:bamToBed [OPTIONS] -i <BAM> ,默认输出的bed 文件有6列

bamToBed -i RNA-seq.bam |head -6
2.5)windowBed
在用户指定的窗口内返回两个BED文件之间的overlap
基本用法:windowBed [OPTIONS] -a <BED> -b <BED> ,默认为上下游1M窗口

2.5.1)默认为上下游1M,例如:


2.5.2)可以通过(-l 和-r)参数来控制上下游,加入不同的window

2.6)closestBed
如果A和B之间没有overlap,则输出与A最近的B的信息
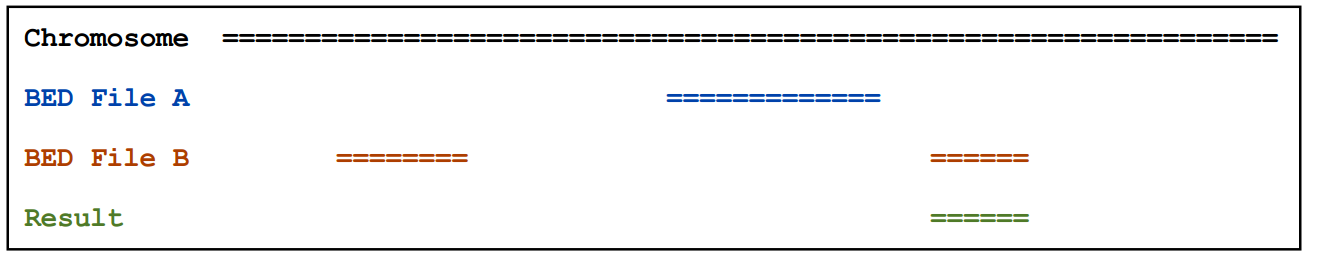
2.7)subtractBed
用处:主要输用于移除重叠部分及间隔区
基本用法:subtractBed [OPTIONS] -a <BED> -b <BED>

默认情况下

2.8)mergeBed
主要用于将重叠的feature合并
基本用法:mergeBed [OPTIONS] -i <BED>

例如:

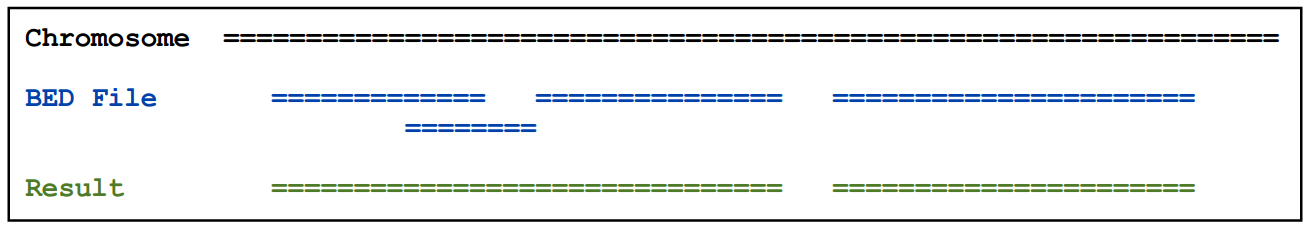 、
、
2.9)coverageBed
主要是统计一个BED文件中要素相对于另一个的覆盖深度和广度
基本用法:coverageBed [OPTIONS] -a <BED> -b <BED>

例如:统计结果解释可以参考下图。
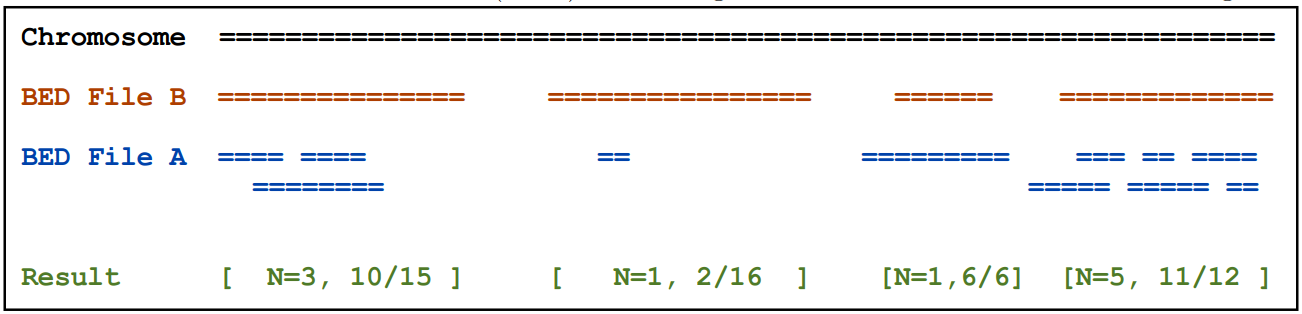
2.10) genomeCoverageBed
对特征文件的覆盖情况进行统计
基本用法:genomeCoverageBed [OPTIONS] -i <BED> -g <GENOME>

2.11)fastaFromBed
用于提取fasta文件中,与bed文件中匹配的序列,形成一个新的fasta文件
基本用法:fastaFromBed [OPTIONS] -fi <input FASTA> -bed <BED> -fo <output FASTA>


2.12)maskFastaFromBed
用于将bed 中指定的位置在fasta中进行mask
基本用法:maskFastaFromBed [OPTIONS] -fi <input FASTA> -bed <BED> -fo <output FASTA>
默认情况下是硬soft,而不是软soft

2.13) shuffleBed
用于置换feture在染色体中的location
shuffleBed [OPTIONS] -i <BED> -g <GENOME>


2.14)sortBed
对bed文件进行排序
基本用法:sortBed [OPTIONS] -i <BED>

默认情况下,对bed文件按照染色体,然后按照起始位置。

2.15)linksBed
为bed 文件建立html
基本用法:linksBed [OPTIONS] -i <BED> > <HTML file>

例如:

2.16)complementBed
输出在bed 文件中不包含的区域
基本用法:complementBed [OPTIONS] -i <BED> -g <GENOME>
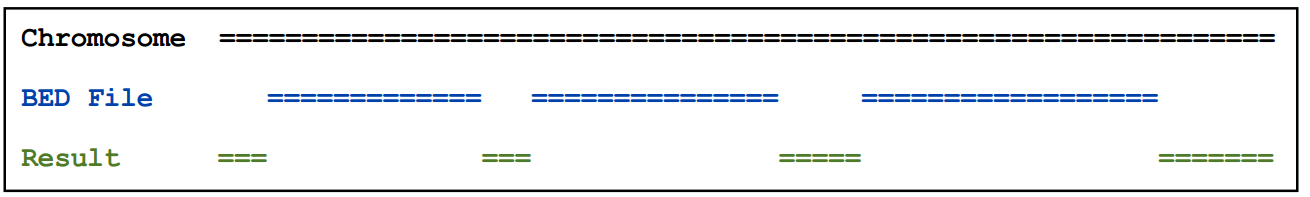
例如


 浙公网安备 33010602011771号
浙公网安备 33010602011771号